3csy
From Proteopedia
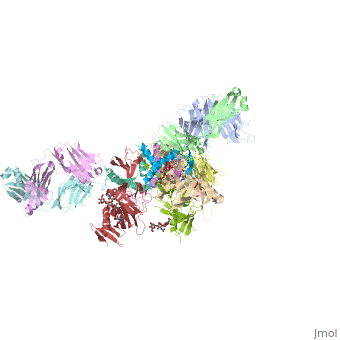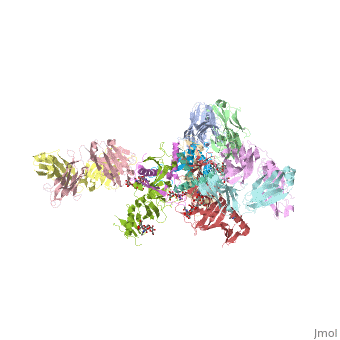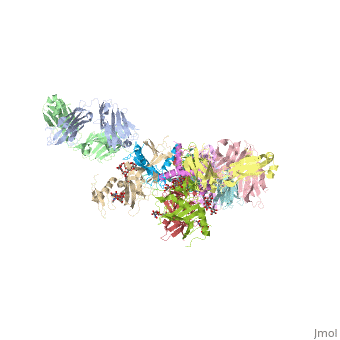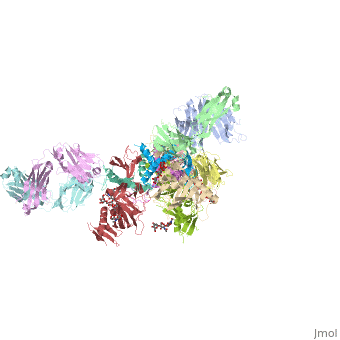
Crystal structure of the trimeric prefusion Ebola virus glycoprotein in complex with a neutralizing antibody from a human survivor
Structural highlights
Function[VGP_EBOZ5] GP1 is responsible for binding to the receptor(s) on target cells. Interacts with CD209/DC-SIGN and CLEC4M/DC-SIGNR which act as cofactors for virus entry into the host cell. Binding to CD209 and CLEC4M, which are respectively found on dendritic cells (DCs), and on endothelial cells of liver sinusoids and lymph node sinuses, facilitate infection of macrophages and endothelial cells. These interactions not only facilitate virus cell entry, but also allow capture of viral particles by DCs and subsequent transmission to susceptible cells without DCs infection (trans infection). Binding to the macrophage specific lectin CLEC10A also seems to enhance virus infectivity. Interaction with FOLR1/folate receptor alpha may be a cofactor for virus entry in some cell types, although results are contradictory. Members of the Tyro3 receptor tyrosine kinase family also seem to be cell entry factors in filovirus infection. Once attached, the virions are internalized through clathrin-dependent endocytosis and/or macropinocytosis. After internalization of the virus into the endosomes of the host cell, proteolysis of GP1 by two cysteine proteases, CTSB/cathepsin B and CTSL/cathepsin L presumably induces a conformational change of GP2, unmasking its fusion peptide and initiating membranes fusion (By similarity). GP2 acts as a class I viral fusion protein. Under the current model, the protein has at least 3 conformational states: pre-fusion native state, pre-hairpin intermediate state, and post-fusion hairpin state. During viral and target cell membrane fusion, the coiled coil regions (heptad repeats) assume a trimer-of-hairpins structure, positioning the fusion peptide in close proximity to the C-terminal region of the ectodomain. The formation of this structure appears to drive apposition and subsequent fusion of viral and target cell membranes. Responsible for penetration of the virus into the cell cytoplasm by mediating the fusion of the membrane of the endocytosed virus particle with the endosomal membrane. Low pH in endosomes induces an irreversible conformational change in GP2, releasing the fusion hydrophobic peptide (By similarity). GP1,2 mediates endothelial cell activation and decreases endothelial barrier function. Mediates activation of primary macrophages. At terminal stages of the viral infection, when its expression is high, GP1,2 down-modulates the expression of various host cell surface molecules that are essential for immune surveillance and cell adhesion. Down-modulates integrins ITGA1, ITGA2, ITGA3, ITGA4, ITGA5, ITGA6, ITGAV and ITGB1. GP1,2 alters the cellular recycling of the dimer alpha-V/beta-3 via a dynamin-dependent pathway. Decrease in the host cell surface expression of various adhesion molecules may lead to cell detachment, contributing to the disruption of blood vessel integrity and hemorrhages developed during Ebola virus infection (cytotoxicity). This cytotoxicity appears late in the infection, only after the massive release of viral particles by infected cells. Down-modulation of host MHC-I, leading to altered recognition by immune cells, may explain the immune suppression and inflammatory dysfunction linked to Ebola infection. Also down-modulates EGFR surface expression (By similarity). GP2delta is part of the complex GP1,2delta released by host ADAM17 metalloprotease. This secreted complex may play a role in the pathogenesis of the virus by efficiently blocking the neutralizing antibodies that would otherwise neutralize the virus surface glycoproteins GP1,2. Might therefore contribute to the lack of inflammatory reaction seen during infection in spite the of extensive necrosis and massive virus production. GP1,2delta does not seem to be involved in activation of primary macrophages (By similarity). [VGP_EBOZM] GP1 is responsible for binding to the receptor(s) on target cells. Interacts with CD209/DC-SIGN and CLEC4M/DC-SIGNR which act as cofactors for virus entry into the host cell. Binding to CD209 and CLEC4M, which are respectively found on dendritic cells (DCs), and on endothelial cells of liver sinusoids and lymph node sinuses, facilitate infection of macrophages and endothelial cells. These interactions not only facilitate virus cell entry, but also allow capture of viral particles by DCs and subsequent transmission to susceptible cells without DCs infection (trans infection). Binding to the macrophage specific lectin CLEC10A also seem to enhance virus infectivity. Interaction with FOLR1/folate receptor alpha may be a cofactor for virus entry in some cell types, although results are contradictory. Members of the Tyro3 receptor tyrosine kinase family also seem to be cell entry factors in filovirus infection. Once attached, the virions are internalized through clathrin-dependent endocytosis and/or macropinocytosis. After internalization of the virus into the endosomes of the host cell, proteolysis of GP1 by two cysteine proteases, CTSB/cathepsin B and CTSL/cathepsin L presumably induces a conformational change of GP2, unmasking its fusion peptide and initiating membranes fusion.[1] [2] [3] [4] [5] [6] [7] [8] GP2 acts as a class I viral fusion protein. Under the current model, the protein has at least 3 conformational states: pre-fusion native state, pre-hairpin intermediate state, and post-fusion hairpin state. During viral and target cell membrane fusion, the coiled coil regions (heptad repeats) assume a trimer-of-hairpins structure, positioning the fusion peptide in close proximity to the C-terminal region of the ectodomain. The formation of this structure appears to drive apposition and subsequent fusion of viral and target cell membranes. Responsible for penetration of the virus into the cell cytoplasm by mediating the fusion of the membrane of the endocytosed virus particle with the endosomal membrane. Low pH in endosomes induces an irreversible conformational change in GP2, releasing the fusion hydrophobic peptide.[9] [10] [11] [12] [13] [14] [15] [16] GP1,2 mediates endothelial cell activation and decreases endothelial barrier function. Mediates activation of primary macrophages. At terminal stages of the viral infection, when its expression is high, GP1,2 down-modulates the expression of various host cell surface molecules that are essential for immune surveillance and cell adhesion. Down-modulates integrins ITGA1, ITGA2, ITGA3, ITGA4, ITGA5, ITGA6, ITGAV and ITGB1. GP1,2 alters the cellular recycling of the dimer alpha-V/beta-3 via a dynamin-dependent pathway. Decrease in the host cell surface expression of various adhesion molecules may lead to cell detachment, contributing to the disruption of blood vessel integrity and hemorrhages developed during Ebola virus infection (cytotoxicity). This cytotoxicity appears late in the infection, only after the massive release of viral particles by infected cells. Down-modulation of host MHC-I, leading to altered recognition by immune cells, may explain the immune suppression and inflammatory dysfunction linked to Ebola infection. Also down-modulates EGFR surface expression.[17] [18] [19] [20] [21] [22] [23] [24] GP2delta is part of the complex GP1,2delta released by host ADAM17 metalloprotease. This secreted complex may play a role in the pathogenesis of the virus by efficiently blocking the neutralizing antibodies that would otherwise neutralize the virus surface glycoproteins GP1,2. Might therefore contribute to the lack of inflammatory reaction seen during infection in spite the of extensive necrosis and massive virus production. GP1,2delta does not seem to be involved in activation of primary macrophages.[25] [26] [27] [28] [29] [30] [31] [32] Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedEbola virus (EBOV) entry requires the surface glycoprotein (GP) to initiate attachment and fusion of viral and host membranes. Here we report the crystal structure of EBOV GP in its trimeric, pre-fusion conformation (GP1+GP2) bound to a neutralizing antibody, KZ52, derived from a human survivor of the 1995 Kikwit outbreak. Three GP1 viral attachment subunits assemble to form a chalice, cradled by the GP2 fusion subunits, while a novel glycan cap and projected mucin-like domain restrict access to the conserved receptor-binding site sequestered in the chalice bowl. The glycocalyx surrounding GP is likely central to immune evasion and may explain why survivors have insignificant neutralizing antibody titres. KZ52 recognizes a protein epitope at the chalice base where it clamps several regions of the pre-fusion GP2 to the amino terminus of GP1. This structure provides a template for unravelling the mechanism of EBOV GP-mediated fusion and for future immunotherapeutic development. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor.,Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO Nature. 2008 Jul 10;454(7201):177-82. PMID:18615077[33] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. See Also
References
| ||||||||||||||||||||||