UMass Chem 423 Student Projects 2011-2
From Proteopedia
Spring 2011 Chem423 Team Projects: Understanding Drug Mechanisms Instructions posted here: Student Projects for UMass Chemistry 423 Spring 2011
Student projects continued below.
Inna Brockman, Robert Nathan, Sarena Horava, Nick Cadirov - p38 kinase
David Peltier, Donald Einck, Ethan Leighton, Chris Coakley - Rituximab Fab
Max Moulton, Sally Stras, Jordan Schleeweis, Anh Huynh -- HIV Reverse Transcriptase
Lyes Khendek, Paul Breslin, William Rowley, Joe Perito, Ashley Rivera - DNA G-Quadruplex
Contents |
p38 kinase
| |||||||||
| 1a9u, resolution 2.50Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligands: | |||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, PDBsum, RCSB | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
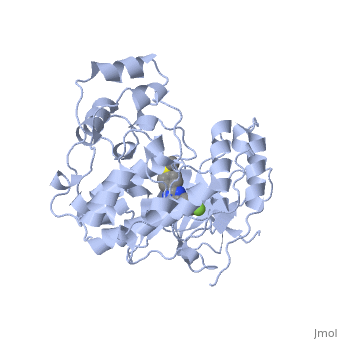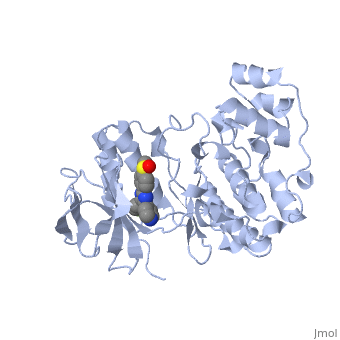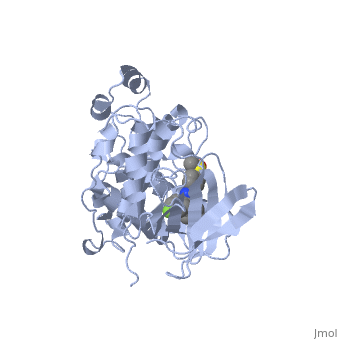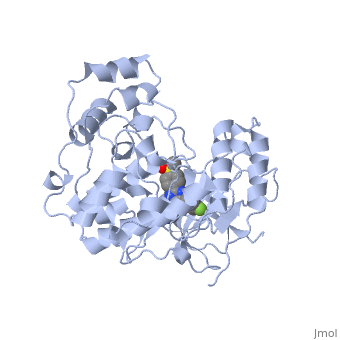

P38 kinase belongs to one of the four subgroups of mitogen-activated protein (MAP) kinases. MAP kinases respond to extracellular stimuli by a signaling cascade leading to intracellular responses. Therefore, MAP kinases function to regulate fundamental cellular processes [1-5].
The p38 subgroup consists of four isoforms: p38α, p38β, p38γ, and p38δ [1-3]. Of these isoforms, p38α and p38β have the most similar amino acid sequences and both forms are expressed in most cell types. P38γ is mostly found in skeletal muscle, while p38δ is only found in the skin, small intestine, pancreas, and kidney [1, 2]. Since p38α was first discovered, most publications focus on this isoform and refer to p38α as p38 [2]. However, all four isoforms have the Thr-Glu-Tyr dual phosphorylation site in the regulatory loop. The substrate specificity of p38 is controlled Glu residue in this dual phosphorylation motif and the length of the loop [1]. This specificity is important for the signal cascade generated in response to stimuli.
Operating as a signal transduction mediator, p38 is activiated by both stress and mitogen stimuli. Environmental stress, particularly UV radiation and osmotic shock, cause an increase activity level of p38. Also, p38 is activated by pro-inflammatory cytokines, especially tumor necrosis factor (TNF) and interleukin-1 (IL1) [3]. However, activation of p38 depends on both the stimulus and the cell type [1]. Dual phosphorylation on the Thr and Tyr is necessary for p38 activation. This dual phosphorylation motif is common in all members of the MAP kinase family. Upstream kinases, which are the MAP kinase kinases (mkks), are responsible for p38 activation [1-3]. Due to selective activation, each p38 isoform requires distinct mkks. Further upstream activators of the MKK/p38 pathway are widely diversified. This cascade accounts for the various stimuli that lead to activating the p38 pathway [1]. Dephosphorylation by dual phosphatases is responsible for the major of the downregulation of p38 [1,3].
The activation of p38 pathway leads to the activation of downstream substrates, such as protein kinases and transcription factors. The p38 pathway regulates close to a hundred genes. P38 is associated with the expression of many cytokines, transcription factors, and cell surface receptors [1]. Various proteins that control transcription and translation are targeted, either directly or indirectly, by p38 kinases. Biological results of p38 activation include inflammation, apoptosis, cell cycle, and cell differentiation [1-3]. However, the role of p38 is specific to cell type [1].
The role of the p38 pathway in cellular inflammation places p38 as a key therapeutic target for inflammatory diseases, cancer, and other diseases. Therefore, p38 inhibitors are key therapeutic agents for the treatment of such diseases. Pyridinyl imidazoles, especially SB203580 (ligand shown in the Jmol diagram), inhibit the catalytic activity of p38 by binding to the ATP site [5]. The ATP binding site provides specificity necessary for highly selective pyridinyl imidazole inhibitors. The inhibitors for p38 do not bind to structurally similar MAP kinases. Other structural factors, such an unique pocket in p38 for the fluorophenyl ring of some pyridinyl imidazole inhibitors, contribute to the selectively of the inhibitor [4]. SB203580 and related inhibitors binds with about equal affinity to both the activated and inactivated forms of p38 kinase. Therefore, binding of the inhibitor can lock p38 into an inactivated conformation.
Gleevec is a brand name drug that targets p38. Gleevec, which is imatinib mesylate, is an inhibitor of p38. Imatinib mesylate, chemically designated as 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-phenyl]benzamide methanesulfonate [6], is structurally similar to SB203580, 4-[5-(4-fluoro-phenyl)-2-(4-methanesulfinyl-phenyl)-3h-imidazol-4-yl]-pyridine [7]. Gleevec is used to treat certain cancers, including chronic myeloid leukemia, gastrointestinal stromal tumors, and myelodysplastic/myeloproliferative diseases. Gleevec inhibits p38, preventing the proliferation of cancer cells [6].
Overall Structure
Kinase is a single 351 amino acid polypeptide chain made up of 10 alpha helixes and 10 beta strands. Kinase p38 is composed of two domains. The first is a 135 residue N-terminal domain and the second a 225 residue C-terminal domain. The beta strands in light blue form antiparallel beta sheets and are located mainly towards the N-terminus while the alpha helixes in green are located mainly towards the C-terminus. In this rainbow representation of the N and C termini are found at the top of the protein. The catalytic site where the drug binds is located between the two domains.
Drug Binding Site
The p38 kinase-SB2 complex binding chemistry is analyzed in this section. The is connected to the binding site by a hydrogen bond with Met109 and the cyclopropylmethyl group binds to the phosphate-binding ribbon in a depression formed by Val30 and Val38. The complex is also stabilized by contributing from Lys53 and Val105. The conformation of the phosphate-binding ribbon of the p38 kinase changes significantly in order to bind with SB2.
Additional Features
Kinase p38 has a between (also known as domains), which the inhibitors can bind. Blocking p38 kinase may be an valuable way of treating many inflammatory diseases. The pyridinylimidazole inhibitors bind in the ATP binding site, making it competitive against ATP. There has also been observed an allosteric binding site in the Asp-Phe-Gly motif in the active site on p38 kinase for a “diaryl urea class of highly potent and selective inhibitors”. These inhibitors have a new binding mechanism, which does not directly compete with . BIRB 796 is an inhibitor, which has a 12,000-fold increase in binding affinity compared to the previous inhibitor. Structural changes that made such an increase in binding affinity are: methyl substituent on pyrazole ring replaced with tolyl group, chlorophenyl group replaced with naphthyl moiety, and addition of ethoxymorpholine subsituent on paththyl ring. Tolyl has hydrophobic interactions with with side chains of Glu 71 side chain. Glu 71 has one hydrogen bond with the NH on urea. This new conformation is better for supporting the hydrophobic interactions on the tolyl group with the inhibitor. Information about the structure of p38 kinase and its interaction with inhibitors is useful for research about reducing inflammation and diseases such as rheumatoid arthritis. New inhibitors are being designed an optimized to help fight inflammation and other diseases such as rheumatoid arthritis.
Credits
Introduction - Sarena Horava
Overall Structure - Robert Nathan
Drug Binding Site - Nick Cadirov
Additional Features - Inna Brockman
References
1. Ono, K.; J. Han, The p83 signal transduction pathway activation and function, Cellular Signalling 12 (2000) 1-13.
2. Min, L.; B. He; L. Hui, Mitogen-activated protein kinases in hepatocellullar carcinoma development, Seminars in Cancer Biology 21 (2011) 10-20.
3. Raingeaud, J; S. Gupta, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine, The Journal of Biological Chemistry 270 (1995) 7420-7426.
4. Wang, Z.; Canagarajah, et al. Structural basis of inhibitor selectivity in MAP kinases, Structure 6 (1998) 1117-1128.
5. Young, P. R.; M. M. McLaughlin, et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site, The Journal of Biological Chemistry 272 (1997) 12116-12121.
6. Gleevec http://www.rxlist.com/gleevec-drug-center.htm
7. Ligand Summary for SB2 http://www.pdb.org/pdb/ligand/ligandsummary.do?hetId=SB2&sid=1A9U
http://www.pdb.org/pdb/explore/explore.do?structureId=1A9U
S. Pav, D. M. White, S. Rogers, K. M. Crane, C. L. Cywin, W. Davidson, J. Hopkins, M. L. Brown, C. A. Pargellis & L. Tong. (1997). Crystallization and preliminary crystallographic analysis of recombinant human p38 MAP kinase. Protein Science, 6, 242-245.
L. Tong, S. Pav, D. M. White, S. Rogers, K. M. Crane, C. L. Cywin, M. L. Brown & C. A. Pargellis. (1997). A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nature Struct. Biol. 4, 311-316.
C. Pargellis, L. Tong, L. Churchill, P.F. Cirillo, T. Gilmore, A.G. Graham, P.M. Grob, E.R. Hickey, N. Moss, S. Pav & J. Regan. (2002). Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nature Struct. Biol. 9, 268-272.
J. Regan, S. Breitfelder, P. Cirillo, T. Gilmore, A.G. Graham, E. Hickey, B. Klaus, J. Madwed, M. Moriak, N. Moss, C. Pargellis, S. Pav, A. Proto, A. Swanimer, L. Tong & C. Torcellini. (2002). Pyrazole urea-based inhibitors of p38 MAP kinase: From lead compound to clinical candidate. J. Med. Chem. 45, 2994-3008.
Drug Binding Site:
Zhulun Wang, Bertram J Canagarajah, Jeffrey C Boehm, Skouki Kassisà, Melanie H Cobb, Peter R Young, Sherin Abdel-Meguid, Jerry L Adams and Elizabeth J Goldsmith. (1998). Structural basis of inhibitor selectivity in MAP kinases. Structure, 6, 1117-1128.
Rituximab Fab
Introduction
Rituximab Fab is used as a prescribed drug to stop transplant rejection, to help regulate autoimmune diseases and to fight cancer. Rituximab has become a key proponent in treatment for cancers such as lymphoma and leukemia and has become the most used chimeric (mouse/human) antibody of its kind. Developed in 1986 by Ivor Royston and Howard Birndorf, Rituximab became FDA approved for clinical use in 1997. Since then Rituximab has become the routine treatment for non-hodgkins lymphoma that otherwise proves to be resistant to certain forms of chemotherapy. Recently, in 2010, Rituximab was approved by the European Commission for use in fighting follicular lymphoma. Because of the wide range of effects that Rituximab causes through its interactions with the CD20 protein, many uses for this antibody have been discovered.
Rituximab is part of a family of antibodies called chimaric monochromal antibodies. These types of antibodies are developed from a single type of parent cell and specifically bind to a single host cell. These types of antibodies do not vary between each other and always interact in the same manner with the chosen cell. Each one of these antibodies is a mixture, chimera, of both rat and human cells both of which play the same role in the host organism. Being chimaric allows for testing on mice before implementation on human cells and combination of human antibodies with mice antibodies makes for simple transitioning between lab testing and clinical testing because the known antibodies have been linked together without making large assumptions with no research.
In brief, Rituximab acts on both benign and malignant lymphocytes, or B-cells. B-cells create antibodies when bombarded with foreign antigens. They are produced in the bone-marrow of humans and when acting normally, help the body fight off disease. If these B-cells are over-produced, under-produced, act abnormally, or are dysfunctional, many different diseases may arise as previously listed. Rituximab acts on the CD20 protein that is found on almost all B-cells. this CD20 protein is unchanged throughout the life of the B-cell.
Multiple proposed mechanisms of action may occur when Rituximab interacts with a lymphocyte (B-cell). The first is known as antibody-dependent cell-mediated cytotoxicity (ADCC) which allows Rituximab to trigger natural cell lysing mechanisms through the use of T-cells and other macrophages. In this proposed mechanism, Rituximab performs as a label to begin cell destruction. Upon further investigation, complement-dependent-cytotoxicity (CDC) became another possible mechanism of action. Through this mechanism, complement proteins are called upon to destroy cell membranes causing overall lysis of the cell. These complement proteins are again naturally occurring, yet usually rely on naturally occurring antibodies to be guided towards non-self antigens. Instead, rituximab acts as the antibody guiding these complements specifically to B-cells. The third and final possible mechanism of action occurs through apoptosis, a more direct route to cell death. Upon initial interaction with the B-cell, the cell triggers programmed cell death. Through this process the cell self-destructs and is immediately destroyed. All three of these mechanisms have been looked into with some uncertainty as to which most likely occurs. Scientists have witnessed many results from treatment such as the shedding of CD23, the down regulation of B-cell receptors, and many other effects that both prove and disprove all three proposed mechanisms.
Rituximab has proven to be a very useful antibody in fighting many human disease, through looking closely at how Rituximab may interact with B-cells, further knowledge about its possible uses will result. This article put together by Chemistry Undergraduate Students at the University of Massachusetts Amherst, will analyze and compile evidence of the chemical mechanisms, binding sites, and applications of Rituximab, a highly used drug in medicine.
Structure
Rituximab is made up of four amino acid chain subunits. Two of these are considered major subunits and two are minor subunits. The two major amino acid sequences are made up of 451 amino acids and the two minor chains contain 213 amino acids. This large protein binds to the surface the surface of B-cells and therefore is exposed primarily to the aqueous external environment. To have stable interactions with the environment, the protein is largely polar, composed almost entirely of beta sheets.
The beta sheets formed in the protein’s native structure form two distinct sides of this protein. Each of these sides is made of woven beta strands that form anti-parallel sheets. The binding site of the protein is found within one of these beta-strand sheets.
Rituximab interacts the surface of its cell membrane through a alpha helix region. This is actually done in two places, where alpha helices of non-polar amino acids are found. Each of these is found at the terminal end of a beta sheet, allowing the active site to bind to the substrate elsewhere on the cell surface.
Each of these structures can be seen in the Jmol native structure found to the right. The red and blue beta strands are the , while the light and dark green strands are the . Each of these has an alpha helix structure, which is shaded in a slightly different color to emphasize its position.
Drug Binding Site
| |||||||||
| 2osl, resolution 2.60Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
Rituximab (Rituximab Fab) is a chimeric antibody with human IgG1 used in the therapy of non-Hodgkin’s B cell lymphomas. This antibody targets B cells by binding to the cell-surface receptor, CD20. CD20 (human B-lymphocyte-restricted differentiation antigen, Bp35) is a hydrophobic transmembrane protein with a molecular weight of approximately 35 kD located on pre-B and mature B lymphocytes. Rituximab has a binding affinity for the CD20 antigen of approximately 8.0 nM, which is similar to the parent murine antibody, 2B8. The amino acid residues alanine (170) and proline (172) within the extracellular loop of CD20 are critical for rituximab binding. Selection of random libraries yielded 2 distinct peptides binding Rituximab: 1 peptide was homologous to alanine (170)-proline(172), the other was assumed to mimic the same epitope.
Of Rituximab Fab, the Fab (fragment antigen-binding) is the region in which the antibody is bound to the antigen. A heavy and light chain domain located at the terminal end of the monomer shapes the binding site of Rituximab Fab. Each one of these chains (heavy and light) has a constant and variable portion. The constant chain is identical in all antibodies; the variable chain is unique to each specific B-cell antibodies. The heavy chains of Rituximab Fab are made up of approximately 451 amino acids and the light chains by approximately 213 amino acids.
Rituximab can begin complement activation by binding of Clq to the Fc region (region of an antibody that interacts with cell surface receptors) of an antibody when inducing complement-mediated cell lysis. C1q is a 400kDa protein split into 3 subunits. Each subunit consists of Y-shaped triple peptide helices joined at the stem, forming a globular non-helical head at its end. The helical components of the structure contain varied strands of Glycine, proline, isoleucine and/or hydroxylysine. The globular heads of the structure (along with two serine proteases, Clr and Cls) are then responsible for the multivalent attachment of the C1q structure; forming the complex C1 which triggers the compliment cascade that performs cellular lysis.
Additional Effects
Rituximab is used quite frequently to treat dysfunctional leukemias and lymphomas. Rituximab works extremely well to treat these diseases because of the CD20 binding site. This treatment can also lead to an increase in the number of circulating CD20+ B cells. Rituximab has also shown to be effective in the treatment of multiple sclerosis, rheumatoid arthritis, and anemias. However while effectiveness has been proven, there are also concerns about the safety of the treatment. There is current research being conducted in Norway which will research the effectiveness of rituximab to treat chronic fatigue syndrome. Rituximab is also being used to manage the recipients of kidney transplants. All of these treatments are because of the CD20 binding sites.
Credits
Introduction: David Peltier
Structure: Donald Einck
Drug Binding Site: Ethan Leighton
Additional Effects: Chris Coakley
Citations: Chris Coakley
All Green Screen Effects: David Peltier
Citations
- Sieber, S, G Gydnia, W Roth, B Bonavida, and T Efferth. "Combination treatment of malignant B cells using the anti-CD20 antibody rituximab and the anti-malarial artesunate." Int J Oncol. 35.1 (2009): 149-158. Print.
- RITUXAN® (Rituximab) full prescribing information, Genentech, Inc., 2008
- DiJulio JE. Monoclonal antibodies: overview and use in hematologic malignancies. In: Rieger PT, ed.Biotherapy: A Comprehensive Overview. 2nd ed. Sudbury, Mass: Jones and Bartlett Publishers; 2001:283-316.
- Maloney DG, Smith B, Rose A. Rituximab: mechanism of action and resistance. Semin Oncol. 2002; 29(suppl 2):2-9
- Idusogie, Esohe, Leonard Presta, Helene Santoro, Pin Wong, and Michael Mulkerrin. "Mapping of the C1q Binding Site on Rituxan, a Chimeric Antibody with a Human IgG1 Fc." J Immunol. 164.4 (2000): 4178-4184. Print
- Du, Jiamu, Hao Wang, Chen Zhong, Baozhen Peng, and Melian Zhang. "Structural Basis for Recognition of CD20 by Therapeutic Antibody Rituximab." J Biol Chem. 3.27 (2007): 15073-15080. Print
- Edwards J, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close D, Stevens R, Shaw T (2004). "Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis". N Engl J Med 350 (25): 2572–81.doi:10.1056/NEJMoa032534.PMID 15201414
- Braendstrup P, Bjerrum OW, Nielsen OJ, Jensen BA, Clausen NT, Hansen PB, Andersen I, Schmidt K, Andersen TM, Peterslund NA, Birgens HS, Plesner T, Pedersen BB, Hasselbalch HC. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adult refractory idiopathic thrombocytopenic purpura. Am J Hematol 2005;78:275-80
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB,Long-term responses seen with rituximab in patients with ITP, Community Oncology Vol. 4 No. 2, February 2007:107
- Polyak MJ, Ayer LM, Szczepek AJ, Deans JP (2003). "A cholesterol-dependent CD20 epitope detected by the FMC7 antibody". Leukemia 17 (7): 1384–9
- Monoclonal antibody FMC7 detects a conformational epitope on the CD20 molecule: evidence from phenotyping after rituxan therapy and transfectant cell analyses. 2001
- Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20 Blood 1994 83:435-445
Drug Binding Site Green Screen Taken from This proteopedia page http://www.proteopedia.org/wiki/index.php/Rituximab
HIV Reverse Transcriptase
Group Members: Anh Huynh, Jordan Schleeweis, Max Moulton, Sally Stras
| |||||||||
| 3dlk, resolution 1.85Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligands: | |||||||||
| Gene: | gag-pol (Human immunodeficiency virus type 1 BH10), gag-pol (Human immunodeficiency virus 1) | ||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, PDBsum, RCSB | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
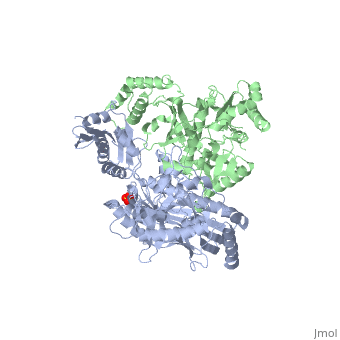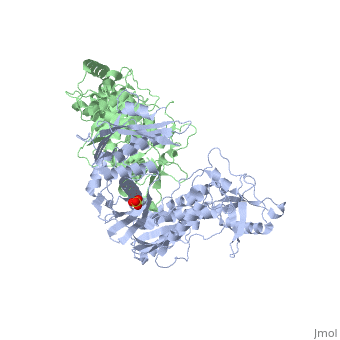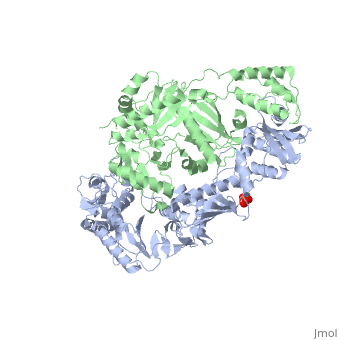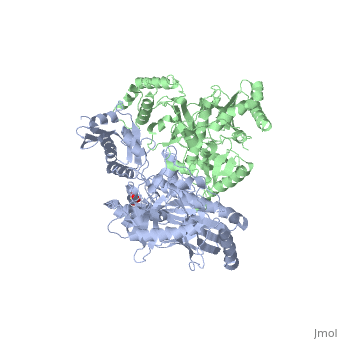
A. INTRODUCTION
HIV-1 reverse transcriptase is required for the creation of double-stranded DNA from mutant single-stranded RNA (hence the term "reverse transcription") [2]. Once mutant single-stranded RNA is transformed into double stranded DNA, the HIV virus can be incorporated into host cells [3]. Anti-HIV drugs frequently utilize mechanisms to inhibit HIV-1 reverse transcriptase. By preventing the proper functionality of HIV-1 reverse transcriptase, the virus's ability to replicate and expand are greatly reduced. These drugs are not always effective because reverse transcription is at high risk for error. DNA mutations due to error facilitate virus resistance to drugs [3]. As a result, anti-HIV drugs must be continuously modified.
B. OVERALL STRUCTURE
HIV-1 RT 3dlk is an asymmetric that contains two subunits, p66 (blue) and p51 (green). The two subunits p66 and p51 consist of 560 and 440 residues respectively. Both subunits consist of alternating at random intervals. There are a total of 32 alpha helicies and 12 beta sheets in the heterodimer. The majority of the beta sheets are constructed in anti-parallel form with a few exceptions. The two subunits are not related in the sense of a similar rotational axis. There are two polymerase domains and one RNase-H domain. The two polymerase domains are the same in sequence but different in conformation. There is one ligand; a that binds to the p66 subunit. The two subunits come together to form the for DNA/RNA. [2]
C. DRUG BINDING SITE
HIV-1 reverse transcriptase (RT) is a primary target for anti-AIDS drugs [2]. Structures of HIV-1 RT have been tested with inhibitor drugs to test the effects of inhibition on RT. These structures have to be specifically engineered in a cost effective way. HIV-1 RT is ligand-independent. The binding sites for possible drugs in HIV-1 RT are in the same location as where the DNA lines up to be replicated [3]. The most successful inhibitor that has been incorporated into HIV-1 RT is called , which binds with the DNA on the hydrophobic inside of the RT. Fragment screening with RT52A/TMC278 crystals is a technique used for finding potential new inhibitors and sites for inhibitors to bind [2]. These tests yielded a site for drug binding interaction called the which is nonengineered unliganded HIV-1 RT [2]. A drug must exhibit noncompetitive inhibition, like Nevirapine, and must bind both the primer-template and dNTP substrates. The specific binding of Nevirapine is a pocket between the beta sheets of the palm (Chain A) and the thumb (highlighted above) subdomains [3]. The drug prevents movements of this thumb subdomain which is needed for the catalytic cycle of HIV. Research continues in this area to determine other mutations in RT that can be engineered for effective drug binding.
D. ADDITIONAL FEATURES
An important part of the HIV reverse transcriptase is an alpha-turn-alpha region of the p66 subunit that has been known to interact with incoming DNA. This portion of the DNA polymerase is the . Is it located in the 'thumb' region and is present in many nucleic acid polymerases and therefore known to assist in the nucleic acid binding [1]. The , located on the p66 subunit, takes part in the cleavage of the RNA/DNA intermediate. The RNase H cleaves the RNA primer from the RNA/DNA hybrid allowing a sythesized DNA to be completed [4].
References
[1] Gotte M, Hermann T, Heumann H, Meier T. The 'helix clamp' in HIV-1 reverse transcriptase: a new nucleic acid binding motif common in nucleic acid polymerases. Nucleic Acids Res.1994 Nov 11;22(22):4625-33.
[2] Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008 Sep;36(15):5083-92. Epub 2008 Aug 1. PMID:18676450 doi:10.1093/nar/gkn464
[3] L. A. Kohlstaedt, J. Wang, J. M. Friedman, P. A. Rice and T. A. Steitz. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 2002 June 26: 256(5056):1783-1790.
[4] Jay F. Davies, II, Zuzana Hostomska, Zdenek Hostomsky, Steven R. Jordan and David A. Matthews. Crystal Structure of the Ribnuclease H domain of HIV-1 reverse transcriptase. Science. 1991 Apr 5;252(5002):89-95. Credits
Introduction -- Anh Huynh
Overall structure --Maxwell Moulton
Drug binding site -- Jordan Schleeweis
Additional features -- Sally Stras
DNA G-Quadruplex
Introduction G-quadruplexes are four single-stranded structures that are formed by nucleic acid sequences rich in guanine. These strcutures are made of a square arrangement of guanines, known as a tetrad, and is stabilized by Hoogsteen face hydrogen bonding. A monovalent cation (either sodium or potassium) in the center of the tetrads also stabilizes these structures. The directions of the strands of the tetrads determine whether the structure is parallel or antiparallel.[3]
The G-quadruplex structures are formed by four single strands of telomeric DNA. Daunomycin binds to the DNA only when DNA is in the G-Quadruplex form and does so by intercalating. In intercalating three molecules of daunomycin fit in between two base pairs and forms hydrogen bonds with the phosphates of the DNA backbone.[2]
For the telomerase enzyme to catalaze synthesis of new telomeric DNA the 3' terminus of the primer DNA needs to be single strand to properly bind with the RNA template of the enzyme. The replication of these telomeric DNA repeats elongates the telomeric DNA and is what prevents apoptosis (cell death) in cancer cells. When the single stranded telomeric DNA is folded into the four-stranded G-Quadruplex it inhibits the telomerase from synthesizing new telomeric DNA. Compounds that promote the formation of the G-Quadruplex are therefore inhibitors of the telomerase enzyme.[2] Telomerase is a tumor-selective target in chemotherapy. The inhibition of telomerase is important because it is over-expressed in about 85% of tumor cells, which leads to the prevention of natural shortening of the telomere and rapid cell growth.[1] This is unlike normal cells, which shorten the telomere sequence after each cell division and leads to the stoppage of cell division (senescence) and then finally apoptosis.[2]
With telomerase inhibition, cancer cells can be tuned back for natural cell death. There must be high binding selectivity towards the intramolecular G-quadruplex structure of telomere over a DNA duplex structure in order to have a G-quadruplex stabilizing agent. [1]
| |||||||
| 1o0k, resolution 1.17Å () | |||||||
|---|---|---|---|---|---|---|---|
| Ligands: | , | ||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||
Structure
Telomeric DNA is mostly double-helical, however; a single strand extends at the 3' terminus. This strand can interact with other strands of Telomeric DNA to form a four stranded quadruplex structure. These quadruplex structures consist of stacked guanine-tetrads that are
apart with a between each G-tetrad. Crystallographic studies have revealed that all strands are parallel in the intermolecular quadruplex formed.[1]
These guanine-tetrads are the repeat motif common to all G-quadrupexes. The G-tetrads consists of a plane of four guanine bases stabilized by hydrogen bonding of the Hoogsteen face. Within any given G-quadruplex there are three to four G-quartets stacked one on top of the other and are further stabilized by pi-pi non-bonded attractive interactions. [4]
The G-quadruplex (G4) has many significant structural differences from double stranded DNA. The two main different features are the four equally sized grooves and a channel of negative electrostatic potential running through the center of the planes of the G-quartets, allowing sodium cations to coordinate between the planes .[2]
Binding Sites
The first crystal structure of a drug bound to a parallel-stranded intermolecular telomeric G4 quadruplex.[1]
The daunomycin layer are tightly packed onto the end of the quadruplex where the daunosamine sugars form H-bonding interactions and/or van der Waals contacts with three of the four quadruplex grooves.
The daunosamine sugar of the first daunomycin is wedged tightly into its groove, and its cationic amine substituent H-bonds to the phosphate oxygens on both sides of the grooves with a distance of 2.81 Å and 2.79 Å. The sugar of the second daunomycin is not as deep in its groove, and H-bonds are found from its cationic amine and exocyclic OH groups to phosphate oxygens on just one side of the groove with a distance of 2.68 Å. Lastly there are no direct H-bonds between the daunosamine of the third daunomycin and the quadruplex. [1]
Additional Features Telomeres shorten in normal somatic cells with each round of replication; this is a result of DNA polymerase's inability to fully replicate the ends of the DNA strands. Eventually the telomere length reaches a critical point that triggers a senescent state at which the cell ceases to replicate, eventually dying. Tumor cells do not suffer this though; they have short, stable telomeres that are replicated by a telomerase enzyme unique to tumor cells. These replicated telomeres are responsible for the immortalization of human cells, leading to tumorigenesis. Inhibition of telomerase leads to natural telomere shortening and eventually cell apoptosis, making telomerase inhibitors an attractive anti-cancer therapy option.[2]
Compounds that have been shown to inhibit the telomerase enzyme are tricyclic aromatic chromophores. Some examples are anthraquinones, fluorenones, and acridines; daunomycin is an acridine. Compounds that show the highest activity has come by substituting the side chains with groups having amidoalkylamino character. The exploration into these synthetic molecules that stabilize the G4 complex have been investigated as potential anti-cancer drugs. Most conventional anti-cancer drugs demonstrate mutual selectivity towards the duplex and quadruplex forms. The distinct structural differences between G2 and G4 allow for the development of new anti-cancer drugs, that are more effective than existing G2 selective drugs.
Credits
Introduction: Joe Perito/Paul Breslin
Overall Structure: Lyes Khendek/William Rowley/Ashley Rivera
Drug Binding Sites: Lyes Khendek
Additional Features: WiIliam Rowley/Paul Breslin
References
[1] Clark GR, Pytel PD, Squire CJ, Neidle S. Structure of the first parallel DNA quadruplex-drug complex. J Am Chem Soc. 2003 Apr 9;125(14):4066-7 http://pubs.acs.org/doi/full/10.1021/ja0297988
[2] Read, M.; Harrison, R. J.; Romagnoli, B.; Taniou, F. A.; Gowan, S. H.; Reszka, A. P.; Wilson, W. D.; Kelland, L. R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors PNAS 2001 Vol. 98, no. 9, pp. 4844-4849
[3] Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S (2006). "Quadruplex DNA: sequence, topology and structure". NAR 34 (19): 5402–5415. doi:10.1093/nar/gkl655. PMC 1636468. PMID 17012276. http://nar.oxfordjournals.org/cgi/content/abstract/34/19/5402.
[4] Shozeb M. Haider, Gary N. Parkinson and Stephen Neidle (2003). "Structure of a G-quadruplex – Ligand Complex." J Mol Biol (2003) 326, 117-125. doi:10.1016/S0022-2836(02)01354-2. http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6WK7-47RC32Y-D&_user=1516330&_coverDate=02%2F07%2F2003&_rdoc=1&_fmt=high&_orig=gateway&_origin=gateway&_sort=d&_docanchor=&view=c&_acct=C000053443&_version=1&_urlVersion=0&_userid=1516330&md5=7b8d86fe20e8957fe1de6a6284e727ab&searchtype=a
