Introduction
Histones
Histones are basic proteins that interact with nuclear DNA to help condense this DNA into chromatin. Nucleosomes are chromatin beads made up of this nuclear DNA wrapped around eight histone proteins, or a histone octamer, in order to fit into the nucleus[1]. The chromatin found inside the nucleus are catagorized into heterochromatin, which are compact and transcriptionally silent, and euchromatin, which are less compact and transcriptionally active[2]. Modifying histones is a type of epigenetics, where changes are made in gene expression while the DNA sequence remains the same. Four different examples of histone modifications include histone acetylation, histone deacetylation, histone methylation and histone demethylation[1].
Histone Deacetylases (HDACs)
HDACs are versatile enzymes that participate in regulating histone-DNA interactions, along with non-histone proteins, the cell cycle, cell differentiation and DNA-histone interactions[3]. Histone acetylation is the reversal process of deacetylation by HDACs, performed by HATs. HDACs perform ε-amino-lysine deacetylation on acetylated lysines, the amount and residue number of which are unclear, within the histone[4].
There are different classes of HDACs based on phylogenetic analysis: Class I (HDACs 1-3 and 8), Class II (HDACs 4-7, 9 and 10), Class III (Sirtuin deacetylases), and Class IV (HDAC 11)
[3]. These different classes share residues that are largely conserved throughout (Figure 1). These conserved residues are either in the active site or involved in substrate binding, as HDACs 1-11 are metalloenzymes with similar reaction mechanisms. They all utilize a zinc ion in their deacetylation
[3].

Figure 1. Conserved Residues Across HDAC Families. Red represents His142 and His143, purple represents Asp178, blue represents His180, pink represents Asp101, yellow represents Tyr306 and green represents Asp267.
One type of HDAC, Histone Deacetylase 8, is an enzyme found in Homo sapiens. HDAC8 decreases gene expression by increasing DNA-histone interactions, condensing the DNA tighter, into the transcriptionally silent heterochromatin[3].
Structure
The structure of HDAC8 was determined via hanging-drop crystallization using a Tyr306Phe mutation (PDB: 2v5w), which was inactive. This allowed the structure to be captured with the substrate bound. The crystallization conditions were: 50 mM Tris–HCl (pH 8.0), 50 mM MgCl2, 10% polyethylene glycol 4000, 2 mM tri(2-carboxyethyl)phosphin and 30 mM glycyl-glycyl-glycine at 22°C. The structure was determined at 2.0Å[3].
HDAC8 exists as a dimer in the crystal structure, is 388 residues long and consists of one eight-stranded parallel surrounded by 11 [3]. HDAC8 is the only functional HDAC that is found to be a single polypeptide instead of being high molecular-weight multi-protein complexes[3].
The Ligand
The bound to the HDAC8 includes an acetyl group, one arginine, one histidine, two lysines and MCM (a Coumarin fluorescence tag, used as a fluorogenic substrate in enzyme kinetics). It was derived from the p53 tumor suppressor gene.
The substrate is held in the binding pocket via several interactions. At the rim of the active site, a conserved makes three hydrogen bonds to two adjacent backbone amides. These interactions force the substrate into a cis conformation. There are also several water molecules that interact with backbone substrate oxygens, assisting the Asp in holding to substrate in the binding pocket during deacetylation reaction. Inhibitors previously crystallized also show these interactions and cis conformation. Mutating Asp101 to an alanine was also shown to abolish enzymatic activity, indicating the importance of the Asp101[3].
The ligand is also held in place via hydrophobic between Phe152, Phe208 and the nonpolar chain of the acetylated lysine. Gly151 also hydrogen-bonds to the amide of the acetyl group, further helping the substrate stay in place[3].
Potassium Ions
There are two potassium ions within HDAC8, which increase the catalytic activity and stability of the enzyme overall. The first is closest to the active site and interacts with active site residues (side chain oxygen of Ser199/Asp176 and the backbone oxygen of Asp176/Asp178/His180/Leu200), with an octahedral geometry. The second potassium ion is about 20 Å from the catalytic center and regulates the enzymatic activity by an allosteric effect[5].
Active Site
The HDAC8 contains features of zinc and serine proteases. Substrates are deacetylated via a classic divalent metal ion mechanism. Zinc pentacoordinates with , the nucleophilic water (making it more acidic) and the carbonyl oxygen of the substrate[3].
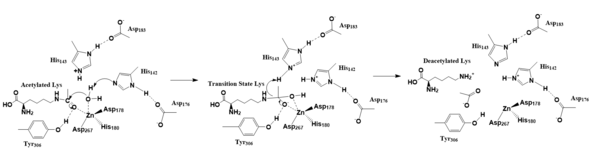
Figure 2. Deacetylation by HDAC8
The zinc ion by withdrawing electron density from the water due to its positive charge, making the water more acidic, therefore a better nucleophile. The zinc ion also coordinates to the carbonyl oxygen of the acetyl group on the lysine, polarizing the carbonyl carbon, making it more electrophilic. There are two present in the active site. His142 deprotonates the water, the first step of the deacetylation (Figure 2). His 142 (stabilized by Asp 176) is closer to the water molecule than His 143 (stabilized by Asp 183), at 2.5Å and 2.8Å, respectively. His143 was also thought to deprotonate the water, however, a mutation done to His143 only reduced activity, not abolished it, showing it is important but not crucial. His143 is instead thought to orient the substrate[3].
The attack by the deprotonated water forces the carbonyl carbon into a tetrahedral transition state. [3] acts as the oxyanion hole, stabilizing this transition state via its protruding -OH group hydrogen-bonding with the negatively charged oxygen. The transition state collapses, and His 143 simultaneously gets deprotonated by the amide of the leaving acetyl group in order to weaken the scissile bond. Breaking this scissile bond results in a neutral lysine and an acetic acid (Figure 2). The deacetylated lysine will be protonated by the hydrogen lost from this acetic acid, providing the hydrogen necessary for lysine to become basic and forming an acetate ion[3].
Inhibition
HDAC Inhibitors
HDAC inhibitors render histone deacetylases inactive. While a variety of these inhibitors are used to target different HDACs, common structural motifs among them include a zinc-binding moiety, a surface recognition or capping group, and a connecting straight chain alkyl, vinyl, or aryl linker. HDAC inhibitors bind and deactivate the HDACs through interactions with the rim amino acids at the active site of the HDAC ()
[6]. The capping group is generally hydrophobic and with these residues (Figure 3)

Figure 3. The anticancer agent Vorinostat is an example of an HDAC inhibitor that is currently undergoing clinical trial.
. Binding occurs via interaction between conserved sections of HDAC active sites and the alkyl, vinyl, or aryl functional groups of the HDAC inhibitors
[7]. ()
[8].
Clinical Application
A variety of HDAC inhibitors are of interest for use as a cancer treatment. HDACs are over expressed in tumor tissue compared to healthy tissue, implying that high amounts of HDAC are reacting with and deacetylating the p53 gene of which the substrate is derived. The p53 gene is a tumor suppressor that, when deacetylated, is rendered inactive due to decreased transcriptional activity and upregulation of oncogenes. The uncontrolled growth of tumors can manifest and lead to cancer. A variety of HDAC inhibitors have been synthesized by modification of the zinc binding moiety or the capping group and tested for development as potential anticancer agents. The rim of amino acids on the catalytic site of HDACs have also been modified to allow for broader specificity of HDAC inhibitors that can bind. Current types of HDAC inhibitors include hydroxamic acids, electrophilic ketones, short chain fatty acids, boronic acid based compounds, benzofuranone, cyclic peptides, and sulfonamide. Of these, two HDAC inhibitors are currently in clinical trials as cancer drugs, known as Vorinostat and Romidepsin[7].



