Overview
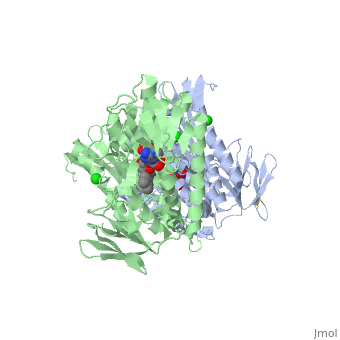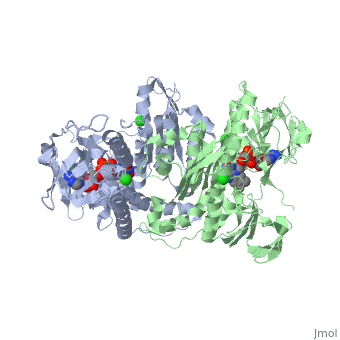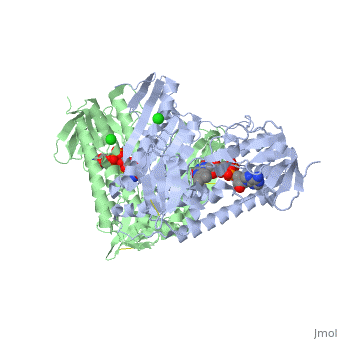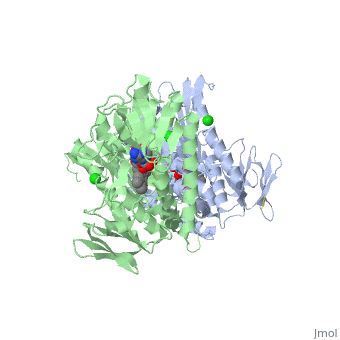
Glutathione reductase belongs to the larger family of flavoezymes, which use a flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN) in catalysis[1]. It is a disulfide oxiodreductase homodimer of 52kD monomers of which, each has four domains[1]:
()
1. NADPH-binding domain (yellow), residues 158-293
2. FAD-binding domain (red) residues 1-157,
3. dimerization domain, residues 365-478.
It is a thermostable protein as it retains 100% of its function up to 65 degrees Celsius attributing to the importance of its function in the cell [2]. It is encoded by a single gene (GeneID:2936, MIM 138300) located on chromosome 8p21.1 [3]. It consists of 13 exons spanning 50kb[3]
For glutathione reductase with nitrotyrosine modification see Nitrotyrosine.
Topology
GSH reductase has a central five-stranded parallel beta-sheet (β1, β2, β3, β7 and β8) [4]. This central β-sheet is surrounded by α-helices 1 and 2 with a crossover connection of a three-stranded antiparallel β-sheet (β4-6) [4].
Compression and Hydride Transfer
Compression has been shown to play a role in enzyme catalysis and X-ray structure of NADPH complex of GSH reductase shows the C4 atom to be only 3.3 Angstrom from the flavin N5 atom[1]. This is closer than one would predict with van der Waals interactions. Compression of the NADPH-bound complex is first evidenced in the decrease in the level of motion of the active site atoms in the complex[1]. Second, it was observed that the van der Waals radii overlapped in the active site, tightly fixing flavin[1]. It is fixed from both sides as flavin N5 to nicotinamide C4 distance is 3.29 Angstrom and the flavin C4a to Cys63-SG distance is 3.29 Angstrom [1]. Third, the flavin ring system shifts in the two reduced structures, with the binding of NADPH pushing the flavin N5 about 0.3 Angstrom towards the thiolate[1]. This provides a structural explanation for how the thiolate-flavin charge transfer intensity is increased by the binding of NADPH [1]. Finally, the last evidence for compression involves the planarity of the nicotinamide group which shows distortion at atom N1 [1].
Reaction
The action of glutathione reductase proceeds through a cyclic series of structures in differing redox states[1]. NADPH binds causing a transient reduction of flavin and this reduced flavin consequently reduces Cys58-Cys63 disulfide bond, forming a short lived covalent intermediate with Cys63[1]. Following this, a stable charge-transfer complex between flavin and the Cys63 thiolate forms[1]. After formation the NADP+ dissociates and is replaced by another NADPH[1]. This is the end of the reductive first half of the mechanism and the oxidative half is initiated upon the binding of GSSG[1]. The Cys58 in glutathione reductase attacks CysI of the GSSG to form a mixed disulfide between the first GS and Cys58[1]. The second GSH is the free to leave and the disulfide bond is reformed between Cys58 and Cys63 of glutathione reductase. Finally, the first molecule of GSH is released[1]. The glutathione reductase is then able to be recycled to allow for the binding of NADPH once again[1].
Function
For proper functioning and prevention of damage to a cell, GSH plays an essential role in preventing oxidative stress in human cells[5]. GSH directly scavenges hydroxyl radicals and singlet oxygens, plays a role as a cofactor in several detoxifying enzymes, participates in amino acid transport through the plasma membrane, and can regenerate important antioxidants such as Vitamins E and C to their reactive forms [5]. The antioxidant capacity of glutathione is linked to the redox state of GSSG/2GSH inside the cell[6]. The function of GSH reductase is to maintain this narrow redox state of high reduced to oxidized ratio of GSH in the cell[7].
Mutation Derived Deficiency
Ultimately, a mutation in the single-copy gene coding for GSH reductase affecting its activity would disrupt the redox state of the GSSG/2GSH in the cell. If GSH were unable to be regenerated from GSSG the cellular environment would become more oxidising, a phenomenon shown to be associated will the onset of cellular apoptosis at a moderate oxidizing environment and necrosis at higher oxidizing cellular environments[8].
Heredity deficiencies of GSH reductase are rare[9]. A deficiency is most often due a genetic mutation and in two related case studies a deletion of the Asp385-Arg478 segment was found [9]. This shorted the protein to 43.8 kDA and once expressed, was likely degraded due to misfolding, leading to a deficiency of GSH reductase[9]. The symptoms and consequences in these genetically related cases were favism, cataracts, and a reduced lifespan of red blood cells[9]. The formation of cataracts in these patients was likely due to UV-induced oxidative damage to the lens of the eye[9].
A second mutation in the GSH reductase gene truncated GSH reductase at Trp287 by changing the TGG codon for Trp287 into a premature TGA stop codon [9]. The folding-initiating helix 11 of residues 439 to 454 is then missing, causing the improper folding and an inactive enzyme [9]. Additionally, Gly330 is exchanged for a GCG codon for alanine [9]. The exchange of glycine to alanine affects catalysis and stability of GSH reductase by disrupting the proper FAD binding necessitating the presence of higher concentrations of FAD to saturate the apoenzyme [9].
Dietary Deficiency
In addition to mutation, an insufficient intake to riboflavin (Vitamin B12) through the diet results in the GSH reductase remaining as an inactive apoenzyme [9]. A deficiency in FAD is usually only found in malnourished populations and these populations make up the more common occurrence of a GSH reductase deficiency[9]. The clinical symptoms of cataracts, favism and reduced lifespans of red blood cells are also seen in dietary deficiency of RSH reductase[9].