Overview
Serum paraoxonases (PONs) or aryldialkylphosphatase are a group of enzymes that play a key role in organophosphate detoxification and in prevention of atherosclerosis. There are three members in this family, PON1, PON2 and PON3, which share 60-70% nucleic acid identity. The most studied enzymes are the two isoenzymes of PON1, which differ in the residue at position 192 (Q/R). The primary activity of PON1 is lactone hydrolysis; however, this enzyme has many other activities. One of the interesting activities is the hydrolysis of organophosphates. PON1 can catalyze a variety of nerve agents such as cyclosarin, soman, etc. Therefore, it is aimed to be a nerve agent scavenger. In addition, PON1 has a role in prevention of atherosclerosis and is found to be attached to the high-density lipoprotein (HDL, “good cholesterol”).
Human PON1 is not stable, and tends to aggregate in the absence of detergents. In addition, it cannot be expressed in bacteria or yeast for protein over-expression, mutagenesis and protein engineering. Therefore, this protein was submitted to directed evolution in order to over-express it in E.coli and to increase its solubility. Family shuffling of four PON1 genes (human, mouse, rabbit and rat) resulted in many variants that could be expressed in E.coli, but only one of them (G2E6-variant) led to quality-diffracted crystals. The recombinant-PON1 (rePON1) G2E6 variant, exhibits 91% homology to the wt rabbit PON1 and 86% homology to the human PON1. This variant exhibits resemble catalytic activity to human PON1.
See also: SsoPox: a natural lactonase with promiscuous phosphotriesterase activities.
Structural features
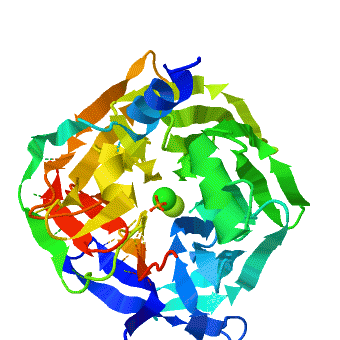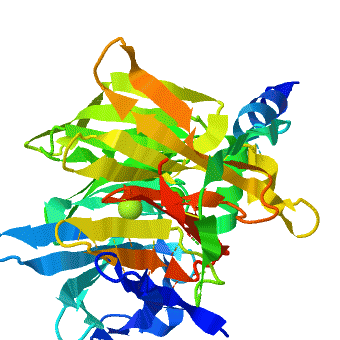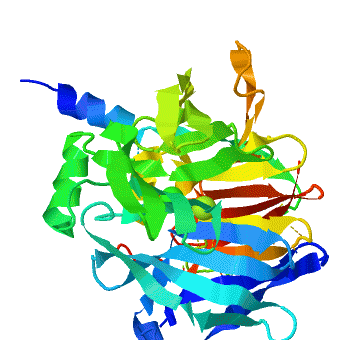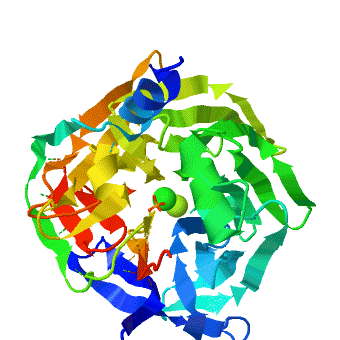
The crystal structure of rePON1 shows[1][2] a β-propeller fold. PON1 has a unique addition to the β-propeller scaffold: three , which are located on the top of the propeller. These helixes are likely to be involved in the anchoring to the HDL particles. In addition, a stabilizing between Cys-42 and Cys-353 was found. The structure of rePON1 resembles that of (PDB 1e1a). Both are six-bladed propellers with each blade consisting of four β-sheets. Moreover, in both structures two can be found in their central tunnel. The calcium atom, which resides at the top of the tunnel, is assigned as the ‘catalytic calcium’ (Ca-1), whereas the other calcium at the central section is assigned as the ‘structural calcium’ (Ca-2). The latter is involved in stabilization of the structure. In addition to the two calciums, there is a , which is bound to Ca-1 in the active site. This phosphate ion is thought to come from the mother liquor. The Loligo vulgaris structure lacks the three α-helixes found in the rePON1 structure.
The catalysis mechanism of organophosphates has yet not been discovered. However, determination of the pH-rate profile of rePON1 proposed participation of a Histidine (His) dyad in the lactonase activity of PON1. In hydrolytic enzymes, His often serves as a base, deprotonating a water molecule, and thus generating the attacking hydroxide ion that produces hydrolysis. The (His-115 and His-134) resides near both Ca-1 and the phosphate ion. The hypothesis is that His-115 acts as a general base to deprotonate a single water molecule, thus generating the attacking hydroxide, while His-134 acts in a proton shuttle mechanism to increase His-115’s basicity. In addition, His-115 was found to have distorted dihedral angles, thing that characterizes many catalytic residues. This observation was supported by site mutation of both His-115 and His-134, which result in a dramatic decrease in both arylesterase and lactonase activity of PON1. Interestingly, the organophosphate hydrolysis activity of these mutations was not affected. Therefore, different loactions in the rePON1 are postulated to have different enzymatic activities in its
.
Catalytic metal ion rearrangements underline promiscuity and evolvability of a metalloenzyme [3]
, especially those playing a catalytic role, exhibit high structural conservation (wildtype serum paraoxonase-1 (PON1) in the presence of either phosphate (PDB: 3sre) or the lactone-analogue 2HQ (PDB: 3srg)). The location of the metal ion and of its ligating residues perfectly superpose, even in distant superfamily members that catalyze different chemical reactions. There exist, however, indications of changes in the configuration of catalytic metals, as part of the catalytic cycle, or upon binding different substrates. Such example is the case of the serum paraoxonase-1 (PON1), in which mutations in the H115 active site residue, induce a (wildtype PON1 is in royalblue, H115W mutant is in magenta and H115Q/H134Q mutant is in salmon). PON1's native activity is the hydrolysis of lipophilic lactones, but it also promiscuously hydrolyzes organophosphates (OPs), particularly paraoxon. It uses different subsets of its catalytic machinery, and different active-site conformations, to catalyze these two reactions. However, the catalytic Ca2+, and its ligating residues are essential for both. is playing a key role in the hydrolysis of lactones. Together with E53, it activates the hydrolytic water for the lactonase activity. Further, mutations to or , reduce the lactonase activity significantly (up to 600-fold). The OP hydrolase activity is, however, enhanced. To gain insight for the structural and mechanistic changes that responsible for this functional transition, the crystal structure of two H115 mutants was determined, and . These crystal structures display major rearrangements of the catalytic metal and of its ligating residues. Specifically, the a 1.8 Å upwards towards the enzyme’s surface, relative to its position in the WT structure. The position of the . Further, the residues coordinating the catalytic Ca2+ are also altered in the mutant structure- the side-chains of . The side-chain of . For . Finally, the side-chains in the vicinity of the mutations also moved, for example for the in order to accommodate the bulkiness of the Trp in position 115. The structural strudies were also complimented with biochemical, mutational and computational analysis that were in good agreement with the structural observations. The computational simulations also suggest a general base catalysis mechanism in which , coordinates and activates the attacking water molecule. These findings, taken together, support the notion that PON1 can accommodate , and that these modes may be used to catalyze different reactions. PON1's native lactonase activity occurs within the , with the location of the catalytic Ca2+ being similar in PON1 and in related enzymes that are highly diverged in their sequences. The promiscuous OPH activity, however, seems to utilize a , and a different mechanism. Alongside the conformational diversity of the protein's backbone and side-chains, metal repositioning may, therefore, contribute to the catalytic versatility of enzymes and to the ease by which new enzymatic functions diverge. The shift in the Ca2+ position, from a rarely populated metal state in the WT to a dominant state in H115W, follows a general model whereby evolution capitalizes on stochastic variations, be they atomic as with PON1's alternative location of the Ca2+, or cellular (e.g., transcriptional noise). Mutations do not create something from nothing. Rather, they shift the distribution such that a marginal, noise phenomenon becomes the norm.
Catalytic versatility and backups in enzyme active sites: The case of serum paraoxanase 1 [4]
The PON1 enzyme has theoretical biological importance as well as application for treatment of neurotoxins. Questions of the origins of enzyme promiscuity or the evolution of protein diversity may be illuminated by PON1's accidental low-level phosphotriesterase activity, and by the unintuitive effect of switching one amino acid in PON1 whereby it changes from a lactonase to a phosphotriesterase. Practically, a potent neurotoxin, "Paraoxon", and therefore a biochemical warfare threat, can be neutralized by phosphotriesterases.
We experimentally solved two critical new PON1 structures. Previously solved in , we have solved PON1 in . While , as expected, there are some key differences. The side-chain of V346 within the active site pocket is , and the side-chains of F347 and H348 in the active site's 'second shell' .
Next, we crystallized , which is a lactone analog. As expected, this structure was also and . We could now see an , most of which had not been seen at either pH 4.5 or 6.5. The first segment of the active site loop, and ,comprises part of PON1's active-site wall. Further, 2HQ's carbonyl oxygen and NH moiety in the apo structure. This overlap supports the notion that both the phosphate ion and 2HQ mimic the binding mode of substrates and/or reaction intermediates. In addition to interacting with the catalytic calcium, 2HQ interacts with the. Importantly, while the bound 2HQ is in contact with the , in the absence of ligand Y71 is either disordered (pH 6.5), or (pH 4.5) .