Introduction
History
Dipeptidyl Peptidase IV's role in the inactivation of incretin hormones was discovered in the 1990s. Animal studies were conducted in the late 1990s, followed by human studies in the early 2000s. The first DPP-IV inhibitors (sitagliptin, vildagliptin, alogliptin, saxagliptin, and linagliptin) were approved starting in 2006, and now serve as monotherapy or add-on to other therapies in a glucose-lowering capacity. [1] Since their approval, there have been multiple long-term trials to continue exploring the long-term effects of these medications. It is known that gliptins directly impact the pancreas, kidney, heart, and vessels. The most investigated thus far is the effects of gliptins on renal and cardiovascular functions. [2] Results of phase II and III trails indicated that gliptins did not harm the cardiovascular system. A meta-analysis implicated two possible beneficial effects for patients treated with these medications: a reduction in cardiovascular effects and a direct renoprotective effect. [3]
Function
DPP-IV, a moonlighting protein, has been implicated in many functions and diseases of the body including glucose metabolism, cardiovascular disease, the stress response, autoimmune diseases (i.e. HIV/AIDS), inflammation, and tumor biology. [4] [5] [6] The active site functions in two ways: it can bind inhibitors and it can truncate substrates. Inhibitors of DPP-IV act via reversible competitive inhibition, binding to the active site but are not degraded, so they remain bound and block the enzymatic activity. Endogenous substrates are truncated by DPP-IV by temporarily forming a covalent bond and ultimately being released in two pieces (the first two residues and the truncated protein).
Structure
Overview
DPP-IV is found in two forms in the body: a membrane bound monomer and a . DPP-IV is more enzymatically active as a dimer than a monomer. Membrane bound DPP-IV is cleaved by a protease between extracellular residues 29-49 to create the blood soluble dimer. Residues have proved to be crucial in the stability of the dimer. The C-terminal loop, the propellor loop interaction sites, and the transmembrane region are all believed to be dimerization interaction sites of DPP-IV, with the transmembrane region playing a significant role in promoting dimerization at any site and maximizing the enzymatic efficiency as a monomer or dimer. [7]
All structural renderings of DPP-IV start at the 39th residue, meaning it does not include the intracellular domain, transmembrane region, and part of the cleavage site. The monomer consists of 4 domains: DPP-IV cleavage stalk, beta propeller, cystine-rich region, and the catalytic domain.
The DPP-IV beta propeller is notable as it differs from all the other enzymes in the dipeptidyl peptidase family. In all other DPPs the beta propeller has ligand gating potential; however, the is an asymmetrical 8 blade propeller that does not function as a ligand gate by rather acts as a binding site which allows DPP-IV to conjugate with Adenosine Deaminase. [8]
The contains 6 cystine residues (C385, C394, C444, C447, C454, C472) that make that play a critical role in the tertiary structure and therefore function of the DPPIV enzyme. [9]
The catalytic domain is where DPP-IV substrates are cleaved at the penultimate point or where inhibitors bind to prevent DPP-IV enzymatic activity.The includes the S1 binding pocket and S2 binding pocket. The S1 binding pocket contains hydrophobic residues (W547, S630, Y631, V656, W659, Y662, Y666, N710, V711 and H740) that interact with either the substrate or inhibitor to keep it within the catalytic domain. The S2 binding pocket contains residues (E205, E206, Y662, S209, R358 and P357) that create hydrogen bonds with either substrate or inhibitor to additionally keep it in place to be cleaved by the catalytic triad. [10] Within these is the , assisted by the oxyanion hole (Y631). [11]
Mechanism
Once a substrate is bound in the active site, DPP-IV utilizes a covalent catalysis mechanism to cleave the substrate at the penultimate position (Figure 2), breaking the scissile bond. D708 of the catalytic triad (S630, H740, D708) pulls electron density from H740, allowing the histidine to pull electron density from S630, making serine a stronger nucleophile. The catalytic triad is assisted in this process by the oxyanion hole (residue Y631), providing stability and keeping the substrate in place. The water molecule attacks the carbonyl carbon, breaking the newly formed covalent bond, and releasing the first two residues of the starting substrate. The active site resets. Figure 1 outlines the mechanism for the truncation of GLP-1 by DPP-IV, breaking the scissile bond at the penultimate position of the protein.
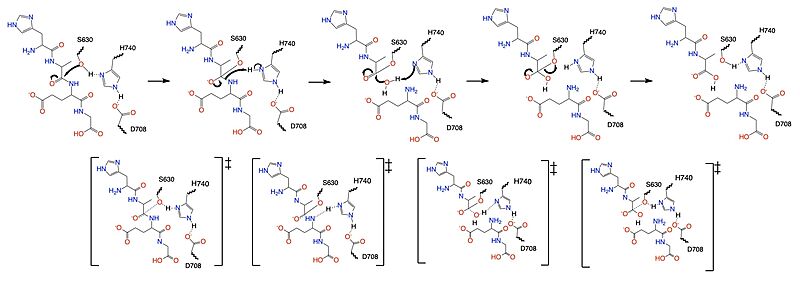
Figure 1. Mechanism for the cleavage of GLP-1 by DPP-IV via the catalytic triad.
Inhibitors
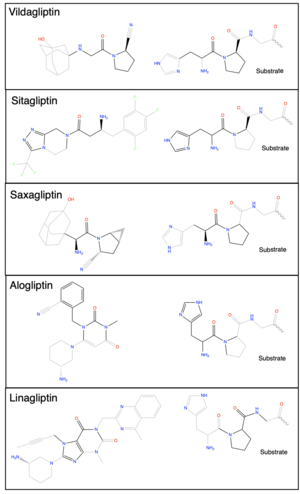
Figure 2. ChemDraw images of the structures of different DPP-IV inhibitors.
DPP-IV inhibition gained traction in the pharmacology field as a reputable therapeutic target in the treatment of type 2 diabetes mellitus (T2DM), following the discovery of GLP-1 and its regulation of insulin signaling. Once experimental trials confirmed DPP-IV to be a key regulator of signaling peptides, including the gliptin peptides, the popularity of DPP-IV inhibition as a treatment of T2DM grew. Clinical trials in the early 2000s confirmed that GLP-1 is able to directly combat T2DM with minimal side effects. The success of the GLP-1 clinical trials gave enormous support for the use of DPP-IV inhibitors as a feasible treatment for T2DM. After the GLP-1 clinical trials, DPP-IV quickly progressed into phase I trial stage. The first DPP-IV inhibitor to be approved in the US by the Food and Drug Administration (FDA) was Sitagliptin in 2006. [12] Sitagliptin was approved as a monotherapy or in combination with Metformin or Thiazolidinedione (TZD) for the treatment of T2DM. After the approval of Sitagliptin in the US the next DPP-IV inhibitor to be approved was in Europe in 2008; however, Vildagliptin [13] is under review in the US. Other currently FDA approved DPP-IV inhibitors include Saxagliptin [14], Linagliptin [11], and Alogliptin (Figure 2). [15]
Gliptins, a class of oral antidiabetic medications, are DPP-IV inhibitors. Each gliptin is a small molecule (≤ 1000 Da). All of the current DPP-IV inhibitors are variations of the same mechanism of inhibition: competitive reversible covalent inhibition. In vivo experimentation has determined that it is crucial to maintain a high inhibitor selectivity as to avoid any inadvertent side effects. The main source of such side effects, as determined by previous trials, resulted from nonspecific inhibition of other isoforms of DPP, namely DPP-8/9. Additionally, DPP-IV possesses auxiliary functions outside of the peptidase catalysis. These functions require that DPP-IV inhibitors are competitive and not allosteric inhibitors to avoid any effect an allosteric inhibitor may have on DPP-IV auxiliary functions. Inhibitors, such as the aforementioned and Vildagliptin, possess a "pseudo N-terminus" functional group whose function is to mimic that of the N-terminus of a DPP-IV substrate, such as GLP-1. The mechanism of covalent inhibition is accomplished through the use of a cyanopyrrolidine group, which forms a covalent bond to DPP-IV. Upon active site binding, the cyanopyrrolidine gorup is sterically located ~2.4 Å from the catalytic S630, to which the carbon of the cyano group forms a covalent bond. The DPP-IV binding pocket consists of the 2 previously mentioned S1 and S2 binding pockets. All DPP-IV inhibitors make varying interactions with these binding pockets and is what allows the inhibitor to have specificity for DPP-IV. In regards to the cyanopyrrolidine group, this binds to the S1 subsite, with the nitrile forming a covalent imidate adduct with the hydroxyl of S630 in the catalytic triad. The imidate nitrogen forms a hydrogen bond with the side chain hydroxyl of Y547. The remaining part, including the adamantane, binds to the S2 subsite, where the carbonyl group forms a hydrogen bond with N710 and the amino group forms salt bridges with E205 and E206. The hydroxyl group of the adamantyl moiety form hydrogen bonds with H126 and S209 via the water molecules. [16] [1] [17]
Medical Relevance
DPP-IV is known to cleave dozens of peptides including, but not limited to, regulatory peptides, neuropeptides, and chemokines. DPP-IV substrates are between 20 and 100 residues long, all of which contain a penultimate proline or alanine, indicating a stereochemical preference (though other penultimate residues are known to be cleaved but with reduced catalytic efficiency)(Figure 3).
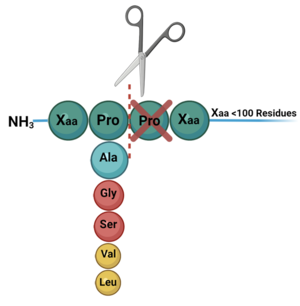
Figure 3. Diagram of the cleavage of substrate at the penultimate position.
Diabetes
Diabetes mellitus is a metabolic disorder, characterized by hyperglycemia, caused by irregularity in insulin secretion, insulin action, or a combination of both. Type 1 diabetes mellitus (T1DM) is characterized by the autoimmune destruction of pancreatic beta-cells, leading to diabetic ketoacidosis. Type 2 diabetes mellitus (T2DM) is characterized by insulin resistance and subsequent deficiency. [18]
(GLP-1) is a 30 amino acid hormone secreted into the gut by intestinal epithelial L-cells. GLP-1 is secreted in response to meal intake and is responsible for stimulating insulin production. GLP-1 binding to the located on the membranes of pancreatic cells results in the upregulation of insulin production and secretion which directly results in the lowering of blood glucose levels. [19]
DPP-IV binds and degrades GLP-1 resulting in heightened blood glucose levels. Insufficient GLP-1 production or signaling in response to meal intake is clinically associated with T2DM and morbidity. Since GLP-1 is a potent regulator of blood glucose levels, and DPP-IV antagonistically regulates GLP-1, this makes DPP-IV inhibitors an excellent candidate for pharmacological therapeutics for T2DM. [20]
Recent studies are demonstrating connections between regulation of Caveolin-1 (Cav-1) and subsequent GLP-1 action. , the principal protein of caveolae, is an integral membrane protein that co-localizes with GLP-1R to regulate receptor trafficking and control the assembly of signaling molecules. Direct interaction of Cav-1 with GLP-1R is required for trafficking and regulation. This interaction occurs with the binding motif in the second intracellular loop of GLP-1R. Two residues in particular, , were proved to be crucial in the binding of Cav-1. Cav-1 also binds to S630 of the DPP-IV catalytic triad thus regulating the activity of DPP-IV and its inhibitors. [21]



