Introduction
Histone Methylation
Human nucleosome particle, pbd code: 5y0c
Histone proteins aid in the packing of DNA for the purpose of compacting the genome in the nucleus of the cell and regulating physical accessibility of genes for transcription. The protein itself is an octamer of core proteins H2a, H2b, H3, and H4, which organize into two heterodimers; H1 and H5 act as linker proteins. About 145-157 base pairs wind around a histone heterodimer core. [1] Modifications to histone core proteins can affect the accessibility of transcription factors in the genome, either promoting or inhibiting transcription. Some of these modifications include methylation/demethylation, acetylation/deacetylation, and ubiquitination/deubiquitination. [2]
Specifically, histone methylation is associated with gene activation. [3] Many domain families fall under the histone methylase family, one of these enzymes being the family, which can target H3, H4, or H2a. Sites known for gene activation are Lysine-4, Lys-36, and Lys-79 on H3; whereas, methylation at Lys-9 and Lys- 27 on H3 and Lys-20 on H4 are known for gene inactivation.[4] Typically, methylation of some of these sites are always present on both active and inactive genes, extra methylations required for activity; specifics of this characteristic depend on site and species of organism. [5] Some tumor related genes such as p53 are site specifically methylated to promote biological function [4], whereas hypomethylation of CpG is linked to tumor genesis. [2] A particular enzyme in the SET7 domain family is lysine methyltransferase, which acts on the histone by adding a methyl group to Lys4 on H3; the addition results in promotion of gene unwinding and gene transcription. [5], [3]
Lysine Methyltransferase (KMT) Structure
The structure of human histone methyltransferase SET7/9 was crystallized using x-ray diffraction at 1.75Å in its product form [5]. SET7/9 was crystallized with its cofactor was in its unmethylated form, S-adenosyl-Lhomocysteine (SAH), and with its product, a ten residue peptide with a methylated lysine at residue 4.
Overall Secondary Structure
Due to the composition of its secondary structure, KMT is an alpha-beta protein [5]. The helical composition includes 3 , with two residing in the SET domain and one in the C-terminal domain. The alpha helices in the SET domain are two turns while the C-terminal helix is by far the largest with 4 turns. There are also 2 in the SET domain which are each one turn. There are 21 total which reside in the N-terminal domain and the SET domain. The beta strands are primarily anti-parallel and multiple antiparallel strands are connected by Type 1 and Type 2 .
The Active Site
The active site and binding pocket of KMT contain residues and shape that optimize catalytic function and stability. First, the lysine of the histone enters the active site via the comprised of Tyr335 and Tyr337. Feeding the histone into the active site is initially difficult; however, once in the active site, the alkyl part of the histone chain is stabilized by the , and polar residues are stabilized by hydrogen bonding interactions on the surface. The Tyr335 and Tyr337 are also essential for stabilization of histone chain via hydrogen bonding. The itself contains the cofactor S-adenosyl methionine (SAM) which donates the methyl group in the reaction. [5] In the active site scene, the structures depict the post-reaction result, where the Lys has been methylated and SAM has been converted to S-adenosyl homocysteine (SAH).
The reaction is catalyzed by Tyr305, Tyr245, carbonyl oxygens of the main chain in residues Ala295 and Ser290. Tyr305 and the carbonyl oxygens of Ala295 and Set 290 coordinate with a water molecule to in turn coordinate with one of the hydrogens off the nitrogen of the lysine, while oxygen of Tyr245 pulls on the other hydrogen of the nitrogen. Both of these actions allow nitrogen to become more nucleophilic and attack the carbon of the methyl group on the SAM, which is attached to a positively charged sulfur. The methyl group is then transferred and charge on the sulfur is resolved; SAM has been converted to SAH. [5]
The C-Terminal Domain
The C-terminal domain of lysine methyltransferase is very important for the catalytic activity of the enzyme. The structures of the serve the role of stabilizing the structures in the in the correct orientation for a reaction in the active site.[5] Hydrophobic interactions in the C-terminal domain are mainly responsible for stabilizing the access channel for the lysine methylation site on histone H3. Residues 337-349 create a that stabilizes the orientation of two tyrosine residues Tyr 335 and Tyr337 that form the lysine access channel. Furthermore, the hydrophobic packing of alpha-helix 3 against beta-sheet 19, specifically , stabilize the orientation of the so that its methyl donating group is oriented toward the lysine access channel. The orientation of the SAM cofactor is further stabilized with its hydrophobic interactions with C-terminal domain residues . [5]
The N-terminal Domain

Figure 1. Clustal alignment of N-terminals
Though a highly conserved region, the of SET7 is notably far from the active site and has not been shown to be involved in enzyme activity or participate in substrate binding. [6] With deletion of the N-terminal domain, studies have shown this modification does not affect SET7 activity. [5] Though not essential for catalytic activity, the N-terminal domain may interact with other small molecules or proteins to act as an allosteric regulator region to the C-terminal domain. [6]
Inhibitors
SET7/9’s structure and function has been studied extensively because of its role in transcription [7]. In the past few years it has been identified to methylate genes involved in multiple diseases; making it a potential candidate for drug inhibition. [8] Two compounds that have been found to inhibit SET7/9 in certain cells in vitro are Sinefungin and Cyproheptadine. Each inhibitor acts on the catalytic center of SET7/9, however their mechanisms of inhibition and possible medical relevancies differ greatly.
Sinefungin
Sinefungin is a potent methyltransferase inhibitor that is a natural nucleoside isolated from the "Streptomyces" species. [8] Also referred to as adenosyl-ornithine, it is the delta (5’ adenosyl) derivative of ornithine and a structural analog of S-adenosylmethionine. Sinefugin is unique because it inhibits where the cofactor binds rather than where the substrate binds like a typical competitive inhibitor. Sinefungin is more stable bound in the active site than SAH due to the ability to create two additional hydrogen bonds to its amine group that are not possible with SAH’s sulfur.
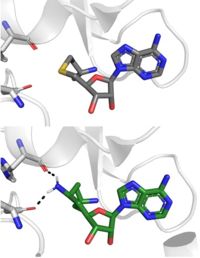
SAH (grey) and Sinefungin (green) in the peptide binding pocket. The nitrogen group of sinefungin makes 2 double bonds to the main chain carbonyls of Arg265 and His293. Sinefungin was created using PDB: 1O9S and mutating the sulfur of SAH
Sinefungin has been used experimentally to inhibit the SET 7/9 protein on peritoneal fibrosis in mice and in human peritoneal mesothelial cells. [8] SET 7/9 is involved in peritoneal fibrosis because it mono-methylates H3K4, which activates the transcription of fibrosis related genes. The administration of Sinefungin to mice in vitro resulted in decreased levels of methylated H3K4 (H3K4me1) protein, as well as suppressed peritoneal cell density and thickening. The decreased levels of H3K4me1 suggest that the methylation of H3K4 was inhibited by Sinefungin, as well as that inhibiting SET7/9 ameliorates peritoneal fibrosis.
Cyproheptadine
Another inhibitor of SET 7/9 is , a clinically-approved anti-allergy drug that was originally developed as a serotonin and histimine. [7] The cyproheptadine-SET 7/9 complex was crystallized via X-ray diffraction at 2.005 Å with methylated cofactor SAM and with cydroheptadine. Unlike Sinefungin, it is a traditional competitor and competitive with the peptide substrates as it binds to the peptide-binding site. When cyproheptadine binds to the substrate site, the nitrogen of the methylpiperdine ring of cyproheptadine forms a hydrogen bond with Thr286 as well as hydrophobic and interactions with the residues surrounding its binding site. The binding of cyproheptadine to the catalytic site causes conformational changes of residue Tyr337, an important residue for the formation of the lysine access channel. This movement subsequently causes a conformational change of the . The conformational change ultimately generates a large hole adjacent to the lysine access channel, as well as the shift of the C-terminal helix.[7]
With the revelation of its inhibitory effects on SET7/9, cyproheptadine was used in vitro to treat breast cancer cells (MCF-7 cells). SET 7/9's non-histone activities include the methylation of the estrogen receptor α (ERα), a nuclear receptor and a transcription factor responsible for estrogen-responsive gene regulation. The expression and transcriptional activity of ERα is involved in the carcinogenesis of 70% of breast cancers, making it a major target for hormone therapy. Researchers found that treating the MCF7 cells with cyproheptadine decreased ERα's expression and transcriptional activity which therefore inhibited the estrogen-dependent cell growth. These findings suggest that cyproheptadine could possibly be repurposed to breast cancer therapy in the future.[7]




