Introduction
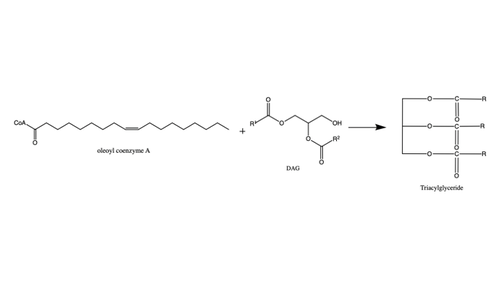
Figure 1: Reaction of oleoyl-CoA with DAG to produce a triglyceride
There are two families of DGAT proteins each with their own distinct cellular functions via synthesis of triacylglycerides from oleoyl-CoA. Diacylglycerol acyltransferase 1 (DGAT1) catalyzes the final and only committed step of triacylclygerol synthesis (Fig. 1). [1] It does this by using diacylglycerol (DAG) and oleoyl CoA as substrates. DGAT1 is located in the membrane of the endoplasmic reticulum and is important for metabolism through its uptake of diacylglycerides and synthesis of triacylglicerides (Fig. 2). [2] This metabolism is involved in intestinal fat absorption, lipoprotein assembly, lactation, and adipose tissue formation [3].
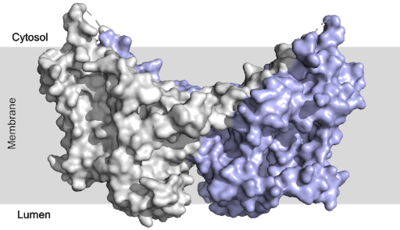
Figure 2: DGAT1 with cytosolic, transmembrane, and luminal domains
Structural Highlights
Conformation
Since DGAT1 is a transmembrane protein, it has 3 distinct regions: cytosolic, transmembrane, and lumenal. Most of the enzyme exists within the membrane with small portions peeking out into the cytosol of the cell or lumen in the surrounding tissue. DGAT1 exists as a homodimer of two identical chains, A and B. The homodimer interface is stabilized in two different ways. First, the transmembrane region is stabilized through large between the TM1 helices on each of the chains. The second is through extensive between the two chains in the cytosolic domain. [2] [4]
Tunnel System
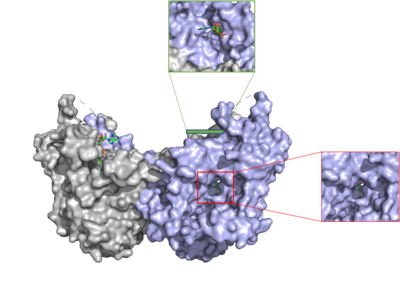
Figure 3: cytosolic tunnel (green) and hydrophobic tunnel (red)
There are two main tunnels that allow the enzymatic activity of DGAT1 to occur. The first is a where the hydrophilic region of the oleyol-CoA binds to the cytosolic face that forms between helices TM7 and TM8. The
CoA region sits at the cytosolic face with the fatty acid chain extending the rest of the way through the enzyme tunnel and leads to the interior of the reaction chamber.
[4] Researchers are currently unsure of specifically what residues are involved in the binding of the oleoyl-CoA to the reaction chamber, but they believe that the hydrogen bonding of the that sits at the cytosolic face of the tunnel. Two residues, His382 and Arg404, are able to form hydrogen bonds to the ligand, and an additional residue, Lys391 forms a salt bridge with the phosphate group of CoA. When hydrophobic residues in the region were mutated, DGAT1 lost all enzymatic activity.
[4] [2]
The second is a that is orthogonal to the cytosolic tunnel. This tunnel is in the transmembrane region of the enzyme which allows lipids in the membrane to easily access the location. [2] Researchers have hypothesized that this tunnel is able to differentiate between DAG, its intended substrate, from other groups that would typically interact with an acyl-CoA, like cholesterol, due to its bent architecture. The bent nature of the tunnel would inhibit more stiff and planar molecules from entering the tunnel and interfering with the activity of the enzyme. [2]
Active Site
The active site of DGAT1 is in the transmembrane region of the enzyme. When it is in its state and no oleoyl-CoA is in its tunnel, the sulfur of Met434 interacts with the catalytic Histidine, His415, which stabilizes the conformation. There are no major conformation changes that take place upon oleoyl-CoA binding into the cytosolic tunnel, however, several key residues change conformation to allow for the entrance of the ligand. In its conformation, His415 hydrogen bonds to Gln465 which stabilizes the Histidine and allows it to be positioned near the thioester bond of the oleoyl-CoA.[2] The His415 interacts with the DAG that enters in a tunnel perpendicular to the oleoyl-CoA. has been hypothesized to be important in holding the DAG in a proper orientation to be able to interact with the oleoyl-CoA and become a triglyceride. [2]
Mechanism
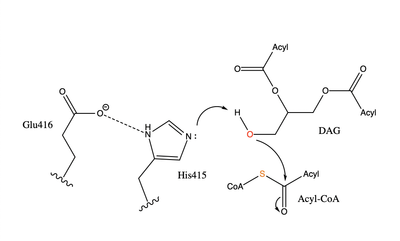
Figure 4: Arrow pushing mechanism of DGAT1 triglyceride synthesis
When a molecule of DAG, or another acyl acceptor, binds into this hydrophobic tunnel, DGAT1 transfers the acyl group on the bound oleoyl-CoA to the DAG to form a triglyceride (Fig. 1). The catalytic histidine deprotonates the hydroxyl group on the C3 of the glycerol backbone (not shown). The deprotonated oxygen then makes a nucleophilic attack on the carbonyl carbon of the Acyl-CoA, the electron density gets shifted up to the oxygen and the tetrahedral oleoyl-CoA-DAG intermediate is formed which is likely stabilized by Gln465.
[4] The electron density then falls back down to the
carbonyl carbon and to the sulfur of the oleoyl-CoA which accepts the added electron density and the bond between the sulfur and carbonyl carbon is broken. (Fig. 4) A proposed interaction between and His415 likely provides enhancement of the reaction by deprotonating His415 which shifts the electron density and helps facilitate the deprotonation of the glycerol backbone. The entrance and binding of the diacylglycerol may cause conformational changes and shifting of the Glu416 to become closer to the His415. Point mutations made to His415 and Glu416 support the hypothesis that it is essential for catalysis in the active site since the enzyme function was completely eliminated when the mutation was made.
[4]
Regulation
Regulation of DGAT1 is thought to be performed by the hydrophilic N-terminal domain which regulates activity based on acyl-CoA/CoA levels. The N-terminal domain contains an intrinsically disordered region and a folded segment. The disordered region has an autoinhibitory function and a dimerization interface, and the folded segment was found to have an allosteric site of acyl-CoA/CoA. When acyl-CoA levels increase, the binding of acyl-CoA with this non-catalytic site initiates allosteric activation. Enzyme activation is prevented until limiting acyl-CoA conditions, which allows CoA to act as a noncompetitive feedback inhibitor. For these reasons, it is proposed that the N-terminal domain of DGAT1 acts as a positive and negative regulator. [5]
Inhibitors
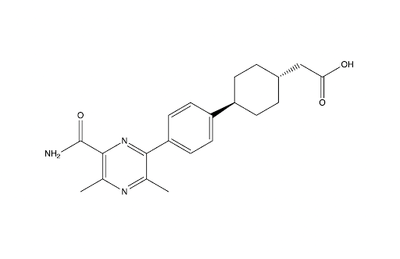
Figure 5: Inhibitor AZD7687
Studies show that reduced DGAT1 function in mice resulted in resistance to obesity when fed a high fat diet and reduced triacylglycerides. This leads to DGAT1 being a potential target for fatty liver disease and hypertriglyceridemia. [4] Inhibiting DGAT1 is thought to decrease obesity and susceptibility to Type 2 Diabetes. The molecule AZD7687 is an inhibitor of DGAT1, though it does not serve as a viable treatment option for obesity or Type 2 Diabetes due to the side effects involved (Fig. 5). As previously mentioned, the inhibition of DGAT1 leads to diarrhea, which proved intolerable for the participants involved in the initial trial of AZD7687. [6] Another molecule, T863, inhibits DGAT1 by acting on the binding site to inhibit DGAT1 (Fig. 6). When given orally, T863 reduces fat absorption and produces results similarly to that of the natural mutations of DGAT1, which again creates doubt for practical use. [7]
Diseases
The two most common and well studied mutations in DGAT1 are an exon 8 deletion mutation and a missense mutation. These mutations both lead to congenital diarrhea.
The first discovery of mutations within the DGAT1 protein were found in chromosome 8 145541756 A→G. This mutation caused exon 8 to be skipped entirely, causing an in-frame deletion of 75 base pairs. [8] This deletion eliminates DGAT1 function, creating a null allele with no DGAT1 expression. This deletion mutation is the more severe mutation, and children with the loss of exon 8 and no DGAT1 function can only handle roughly 4-7% of their consumption to be fat containing calories.
The homozygous recessive L105P missense mutation causes a partial loss in triacylglyceride synthesis. This mutation results in a higher tolerance for percentage of fat intake at around 10% of calories. There are no therapies to restore or increase DGAT1 function in those with mutations, but dietary modifications can assist in minimizing symptoms.
The proper mechanism for how these mutations cause congenital diarrhea is still known. There is a strong hypothesis that DGAT1 lipid substrates from the diet may accumulate in the intestine and cause lipotoxic stress, but this has yet to be proven. [9]






