SARS-CoV-2 spike protein fusion transformation
From Proteopedia
The spike protein of SARS-CoV-2 plays a central role in coronavirus attachment to the ACE2 receptor on host cells, and in getting the RNA genome of the virus into the host cell via fusion of the virus and host cell membranes, initiating infection. You may find it helpful to read SARS-CoV-2 spike protein priming by furin before continuing with the article below.
| |||||||||||
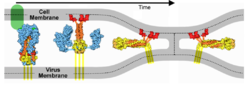



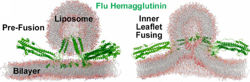

Animations for Slides
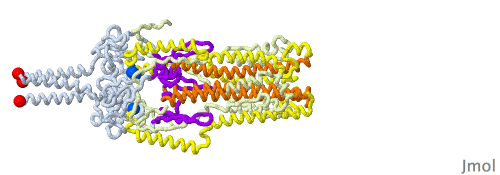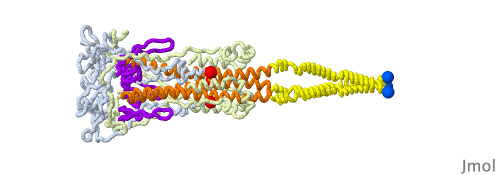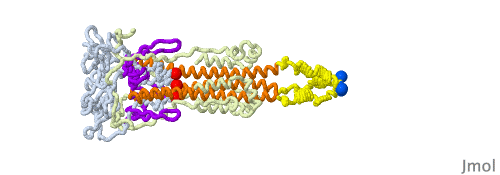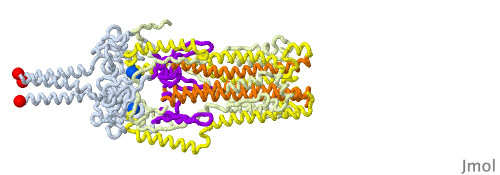
These animations are ready to be dropped into presentation slides (Powerpoint, Google Slides, Libre Office Impress, etc.). As above, the red balls are close to the virus envelope membrane, and the blue balls are close to the host cell membrane. As above, these are linear interpolation morphs from 6xr8 to 6xra.
Solid Fusion Transformation
Download (right click, save link as) 500 px wide solid animation. This shows alpha carbons only, exaggerated in size to 6.2 Å diameter to make a solid object. Please credit Proteopedia.Org in accord with our license, and Cai, Zhang and coworkers[1] for their cryo-EM structures.

Main Chain Fusion Transformation
Download (right click, save link as) 500 px wide main chain animation. This shows a smoothed main chain (backbone) trace. Please credit Proteopedia.Org in accord with our license, and Cai, Zhang and coworkers[1] for their cryo-EM structures.

Alternate Pathway for Transformation
Given that a morph is intended to help you compare two structures and does not portray a realistic transition pathway, there are many pathways we can imagine. Below is another one that has structural elements swing away from the core structure in a hinge motion as they change conformation. Another choice in this morph is to have different timings for the distinct changes to be able to follow them one at a time.
In the morph at full resolution, you can see a large cavity disappearing, the beta sheets acquiring an additional strand from a different subunit (subunits shown in green, blue and red tints), and changes in the core helix bundle along with the formation of the coiled-coil protrusion made of three helices.
Download (right click, save link as) 720 px wide main chain animation. This shows a smoothed main chain (backbone) trace. Please credit Proteopedia.Org in accord with our license, and Cai, Zhang and coworkers[1] for their cryo-EM structures.

Download (right click, save link as) 720 px wide main chain animation. This shows C-alpha atoms as large spheres. Please credit Proteopedia.Org in accord with our license, and Cai, Zhang and coworkers[1] for their cryo-EM structures.

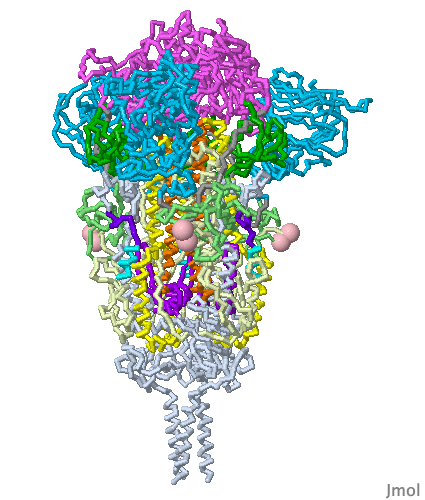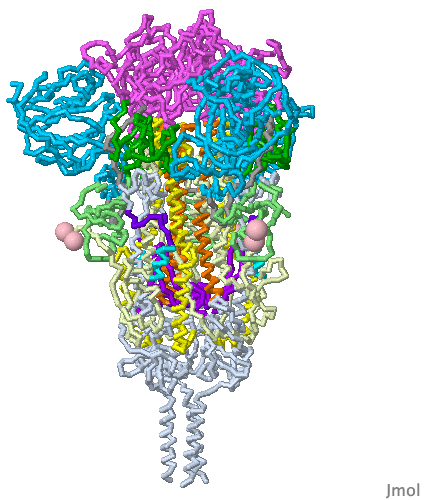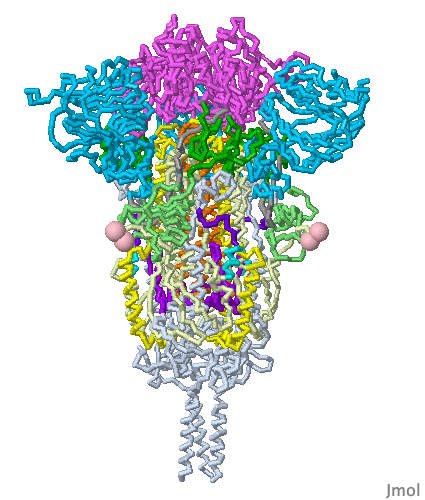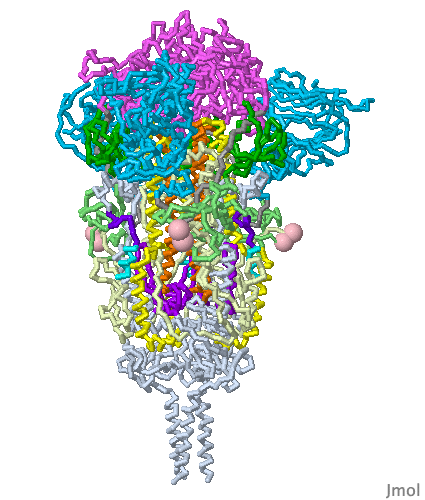
Pre-Fusion Spike Protein
These show SARS-CoV-2 spike protein 6xr8 colored by domain (see color key above). The receptor binding domains are at the top. The bottom extends through parts missing in this model: stem, transmembrane domain, cytoplasmic domain. Pink balls mark the furin cleavage sites that separate S1 from S2. S1 is translucent in the movie at right. Please credit Proteopedia.Org in accord with our license, and Cai, Zhang and coworkers[1] for their cryo-EM structures.
| DOWNLOAD | DOWNLOAD |

| 
|
Other Animations?
If you would like a presentation-ready animation of something else, feel free to contact us.
See Also
- SARS-CoV-2 spike protein priming by furin - the step prior to membrane fusion.
- SARS-CoV-2 spike protein mutations suspected to increase transmission rates.
- SARS-CoV-2 protein S
- Spike protein
- Coronavirus Disease 2019 (COVID-19)
- Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up about 6vsb
- Jmol/Cavities pockets and tunnels
- Cavity programs
Methods
Morphing
The pre-fusion structure 6xr8 was morphed to the post-fusion structure 6xra by linear interpolation, requesting 14 intermediate frames (16 total), using the server provided by Karsten Theis after another method[12] gave unsatisfactory results. To avoid artifactual movement in the morph, prior to morphing, two changes were required in 6xra: (i) the names of chains B and C needed to be swapped (done with SwissPDBViewer), and (ii) the structure needed to be rotated +120° around the Z axis (Jmol "rotateselected" command). The morph was an 11 MB file, which took 25 sec to load into JSmol. Each script took a minimum of 8 sec to complete. To reduce both the bulk of this file and the processing times for JSmol, the alpha carbons were extracted (along with the MODEL and ENDMDL records) by deleting all other lines in the PDB file[13]. The resulting 16 model morph PDB file is Image:Morf-6xr8-6xra-theis-cao.pdb.
Interior Cavities
6zgi was loaded into the Jmol Java application (~11-fold faster than JSmol), and rendered as translucent backbone, each chain a different pastel color. The command
- isosurface minset 100 interior cavity 3.0 10.0
was executed (~45 sec). The numeric parameters in that command were determined by trial and error (see Jmol/Cavities pockets and tunnels). The cavity surface data were saved as Jmol voxel data, and uploaded to Proteopedia as Image:6zgi-cavities.jvxl. The button above reads that file, rather than re-calculating the cavities, in order to display the interior cavity surface data much more quickly. See also Jmol/Cavities pockets and tunnels.
Acknowledgement
Eric Martz thanks Deborah Spitz for a critique that improved this article.
References and Notes
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 Cai Y, Zhang J, Xiao T, Peng H, Sterling SM, Walsh RM Jr, Rawson S, Rits-Volloch S, Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020 Jul 21. pii: science.abd4251. doi: 10.1126/science.abd4251. PMID:32694201 doi:http://dx.doi.org/10.1126/science.abd4251
- ↑ Fan X, Cao D, Kong L, Zhang X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat Commun. 2020 Jul 17;11(1):3618. doi: 10.1038/s41467-020-17371-6. PMID:32681106 doi:http://dx.doi.org/10.1038/s41467-020-17371-6
- ↑ Walls AC, Tortorici MA, Snijder J, Xiong X, Bosch BJ, Rey FA, Veesler D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017 Oct 17;114(42):11157-11162. doi:, 10.1073/pnas.1708727114. Epub 2017 Oct 3. PMID:29073020 doi:http://dx.doi.org/10.1073/pnas.1708727114
- ↑ 4.0 4.1 Hamilton BS, Whittaker GR, Daniel S. Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses. 2012 Jul;4(7):1144-68. doi: 10.3390/v4071144. Epub 2012 Jul 24. PMID:22852045 doi:http://dx.doi.org/10.3390/v4071144
- ↑ 5.0 5.1 5.2 Pabis A, Rawle RJ, Kasson PM. Influenza hemagglutinin drives viral entry via two sequential intramembrane mechanisms. Proc Natl Acad Sci U S A. 2020 Mar 31;117(13):7200-7207. doi:, 10.1073/pnas.1914188117. Epub 2020 Mar 18. PMID:32188780 doi:http://dx.doi.org/10.1073/pnas.1914188117
- ↑ 6.0 6.1 According to Cai, Zhang et al. (their Fig. 4), the initial cut by furin occurs between Arg685 and Ser686 at * in the sequence PRRAR*SVASQ. Thus, S1 is 13-685 (length 673, excluding a 12 residue signal sequence), or 53% of the original chain, leaving S2 as 686-1273, length 588, 47%.
- ↑ The Storymorph Jmol scripts were used to create the interpolation shown in the morph. [1] available on Proteopedia
- ↑ Permission obtained from Gary R. Whittaker August 6, 2020. Permission obtained from David Goodsell August 5, 2020.
- ↑ Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, Skehel JJ, Gamblin SJ. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol. 2020 Jul 9. pii: 10.1038/s41594-020-0468-7. doi:, 10.1038/s41594-020-0468-7. PMID:32647346 doi:http://dx.doi.org/10.1038/s41594-020-0468-7
- ↑ 10.0 10.1 10.2 Romeo A, Iacovelli F, Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020 Jun 18;286:198068. doi: 10.1016/j.virusres.2020.198068. PMID:32565126 doi:http://dx.doi.org/10.1016/j.virusres.2020.198068
- ↑ This quote is from the PubMedCentral version of the Romeo paper, after you click License Information: "Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID-19. The COVID-19 resource centre is hosted on Elsevier Connect, the company's public news and information website. Elsevier hereby grants permission to make all its COVID-19-related research that is available on the COVID-19 resource centre - including this research content - immediately available in PubMed Central and other publicly funded repositories, such as the WHO COVID database with rights for unrestricted research re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for free by Elsevier for as long as the COVID-19 resource centre remains active."
- ↑ Proteopedia's PyMOL morph server was used in both RigiMOL and linear modes, all atoms or only alpha carbon atoms. Rendering these as backbones or traces by Jmol gave broken lines. The reason for backbone breaking was not investigated further.
- ↑ Selecting *.ca in the Jmol Java application and saving a PDB file produced a PDB file with numerous errors. The desired result was obtained with this command in macOS Terminal: sed -e /REMARK/d -e /HETATM/d -e /^ATOM\ \ [\ 0-9][0-9][0-9][0-9][0-9]\ \ [CONS][\ B-Z].*$/d <original.pdb >product.pdb.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}