2pea
From Proteopedia
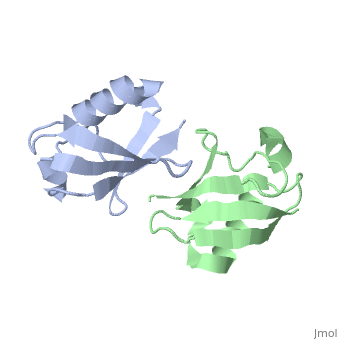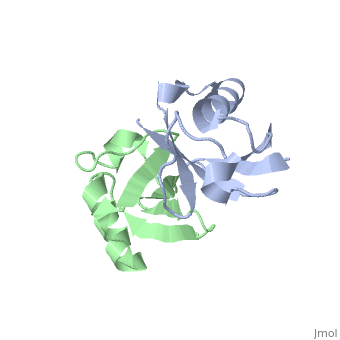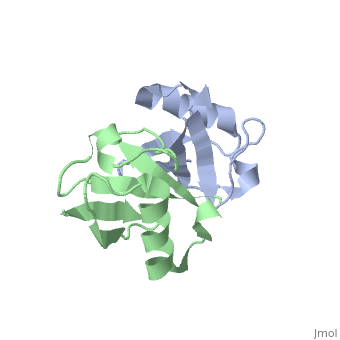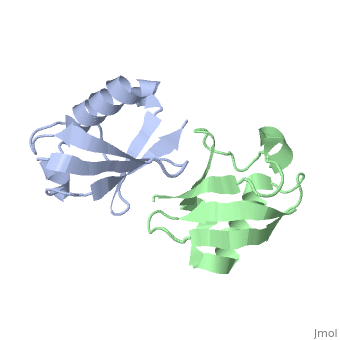
NMR Based Structure of the Closed Conformation of LYS48-Linked Di-Ubiquitin Using Experimental Global Rotational Diffusion Tensor from NMR Relaxation Measurements
Structural highlights
FunctionUBC_HUMAN Ubiquitin exists either covalently attached to another protein, or free (unanchored). When covalently bound, it is conjugated to target proteins via an isopeptide bond either as a monomer (monoubiquitin), a polymer linked via different Lys residues of the ubiquitin (polyubiquitin chains) or a linear polymer linked via the initiator Met of the ubiquitin (linear polyubiquitin chains). Polyubiquitin chains, when attached to a target protein, have different functions depending on the Lys residue of the ubiquitin that is linked: Lys-6-linked may be involved in DNA repair; Lys-11-linked is involved in ERAD (endoplasmic reticulum-associated degradation) and in cell-cycle regulation; Lys-29-linked is involved in lysosomal degradation; Lys-33-linked is involved in kinase modification; Lys-48-linked is involved in protein degradation via the proteasome; Lys-63-linked is involved in endocytosis, DNA-damage responses as well as in signaling processes leading to activation of the transcription factor NF-kappa-B. Linear polymer chains formed via attachment by the initiator Met lead to cell signaling. Ubiquitin is usually conjugated to Lys residues of target proteins, however, in rare cases, conjugation to Cys or Ser residues has been observed. When polyubiquitin is free (unanchored-polyubiquitin), it also has distinct roles, such as in activation of protein kinases, and in signaling.[1] [2] Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedWe present a simple and robust approach that uses the overall rotational diffusion tensor as a structural constraint for domain positioning in multidomain proteins and protein-protein complexes. This method offers the possibility to use NMR relaxation data for detailed structure characterization of such systems provided the structures of individual domains are available. The proposed approach extends the concept of using long-range information contained in the overall rotational diffusion tensor. In contrast to the existing approaches, we use both the principal axes and principal values of protein's rotational diffusion tensor to determine not only the orientation but also the relative positioning of the individual domains in a protein. This is achieved by finding the domain arrangement in a molecule that provides the best possible agreement with all components of the overall rotational diffusion tensor derived from experimental data. The accuracy of the proposed approach is demonstrated for two protein systems with known domain arrangement and parameters of the overall tumbling: the HIV-1 protease homodimer and Maltose Binding Protein. The accuracy of the method and its sensitivity to domain positioning are also tested using computer-generated data for three protein complexes, for which the experimental diffusion tensors are not available. In addition, the proposed method is applied here to determine, for the first time, the structure of both open and closed conformations of a Lys48-linked diubiquitin chain, where domain motions render impossible accurate structure determination by other methods. The proposed method opens new avenues for improving structure characterization of proteins in solution. Structural assembly of multidomain proteins and protein complexes guided by the overall rotational diffusion tensor.,Ryabov Y, Fushman D J Am Chem Soc. 2007 Jun 27;129(25):7894-902. Epub 2007 Jun 6. PMID:17550252[3] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. See AlsoReferences
| ||||||||||||||||||