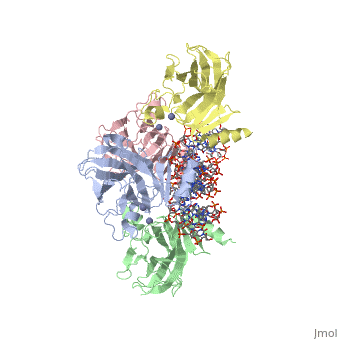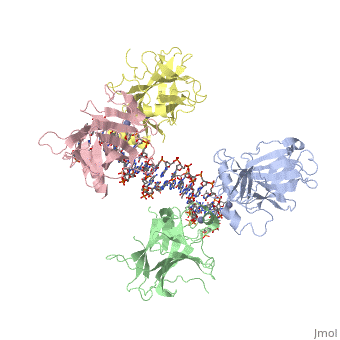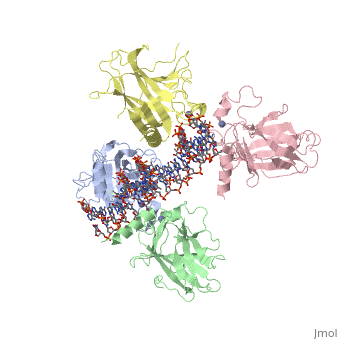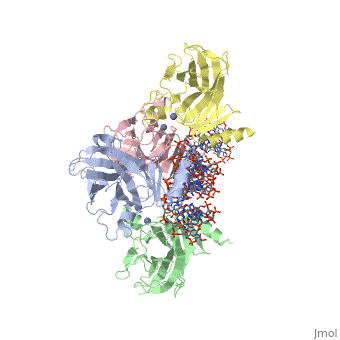
| Structural highlights
Disease
P63_HUMAN Defects in TP63 are the cause of acro-dermato-ungual-lacrimal-tooth syndrome (ADULT syndrome) [MIM:103285; a form of ectodermal dysplasia. Ectodermal dysplasias (EDs) constitute a heterogeneous group of developmental disorders affecting tissues of ectodermal origin. EDs are characterized by abnormal development of two or more ectodermal structures such as hair, teeth, nails and sweat glands, with or without any additional clinical sign. Each combination of clinical features represents a different type of ectodermal dysplasia. ADULT syndrome involves ectrodactyly, syndactyly, finger- and toenail dysplasia, hypoplastic breasts and nipples, intensive freckling, lacrimal duct atresia, frontal alopecia, primary hypodontia, and loss of permanent teeth. ADULT differs significantly from EEC3 syndrome by the absence of facial clefting. Defects in TP63 are the cause of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) [MIM:106260. AEC is an autosomal dominant condition characterized by congenital ectodermal dysplasia with coarse, wiry, sparse hair, dystrophic nails, slight hypohidrosis, scalp infections, ankyloblepharon filiform adnatum, maxillary hypoplasia, hypodontia and cleft lip/palate.[1] Defects in TP63 are the cause of ectrodactyly-ectodermal dysplasia-cleft lip/palate syndrome type 3 (EEC3) [MIM:604292. EEC3 is an autosomal dominant syndrome characterized by ectrodactyly of hands and feet, ectodermal dysplasia and facial clefting.[2] [3] [4] [5] Defects in TP63 are the cause of split-hand/foot malformation type 4 (SHFM4) [MIM:605289. Split-hand/split-foot malformation is a limb malformation involving the central rays of the autopod and presenting with syndactyly, median clefts of the hands and feet, and aplasia and/or hypoplasia of the phalanges, metacarpals, and metatarsals. There is restricted overlap between the mutational spectra of EEC3 and SHFM4.[6] [7] Defects in TP63 are the cause of limb-mammary syndrome (LMS) [MIM:603543. LMS is characterized by ectrodactyly, cleft palate and mammary-gland abnormalities.[8] Note=Defects in TP63 are a cause of cervical, colon, head and neck, lung and ovarian cancers. Defects in TP63 are a cause of ectodermal dysplasia Rapp-Hodgkin type (EDRH) [MIM:129400; also called Rapp-Hodgkin syndrome or anhidrotic ectodermal dysplasia with cleft lip/palate. Ectodermal dysplasia defines a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. EDRH is characterized by the combination of anhidrotic ectodermal dysplasia, cleft lip, and cleft palate. The clinical syndrome is comprised of a characteristic facies (narrow nose and small mouth), wiry, slow-growing, and uncombable hair, sparse eyelashes and eyebrows, obstructed lacrimal puncta/epiphora, bilateral stenosis of external auditory canals, microsomia, hypodontia, cone-shaped incisors, enamel hypoplasia, dystrophic nails, and cleft lip/cleft palate.[9] [10] [11] [12] Defects in TP63 are the cause of non-syndromic orofacial cleft type 8 (OFC8) [MIM:129400. Non-syndromic orofacial cleft is a common birth defect consisting of cleft lips with or without cleft palate. Cleft lips are associated with cleft palate in two-third of cases. A cleft lip can occur on one or both sides and range in severity from a simple notch in the upper lip to a complete opening in the lip extending into the floor of the nostril and involving the upper gum.
Function
P63_HUMAN Acts as a sequence specific DNA binding transcriptional activator or repressor. The isoforms contain a varying set of transactivation and auto-regulating transactivation inhibiting domains thus showing an isoform specific activity. Isoform 2 activates RIPK4 transcription. May be required in conjunction with TP73/p73 for initiation of p53/TP53 dependent apoptosis in response to genotoxic insults and the presence of activated oncogenes. Involved in Notch signaling by probably inducing JAG1 and JAG2. Plays a role in the regulation of epithelial morphogenesis. The ratio of DeltaN-type and TA*-type isoforms may govern the maintenance of epithelial stem cell compartments and regulate the initiation of epithelial stratification from the undifferentiated embryonal ectoderm. Required for limb formation from the apical ectodermal ridge. Activates transcription of the p21 promoter.[13] [14] [15] [16] [17] [18] [19]
See Also
References
- ↑ McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001 Feb 1;10(3):221-9. PMID:11159940
- ↑ Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, Newbury-Ecob R, Hennekam RC, Van Buggenhout G, van Haeringen A, Woods CG, van Essen AJ, de Waal R, Vriend G, Haber DA, Yang A, McKeon F, Brunner HG, van Bokhoven H. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999 Oct 15;99(2):143-53. PMID:10535733
- ↑ Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet. 2000 Jul;67(1):59-66. Epub 2000 Jun 5. PMID:10839977 doi:S0002-9297(07)62432-X
- ↑ van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001 Sep;69(3):481-92. Epub 2001 Jul 17. PMID:11462173 doi:S0002-9297(07)61160-4
- ↑ Akahoshi K, Sakazume S, Kosaki K, Ohashi H, Fukushima Y. EEC syndrome type 3 with a heterozygous germline mutation in the P63 gene and B cell lymphoma. Am J Med Genet A. 2003 Jul 30;120A(3):370-3. PMID:12838557 doi:10.1002/ajmg.a.20064
- ↑ Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet. 2000 Jul;67(1):59-66. Epub 2000 Jun 5. PMID:10839977 doi:S0002-9297(07)62432-X
- ↑ van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001 Sep;69(3):481-92. Epub 2001 Jul 17. PMID:11462173 doi:S0002-9297(07)61160-4
- ↑ van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001 Sep;69(3):481-92. Epub 2001 Jul 17. PMID:11462173 doi:S0002-9297(07)61160-4
- ↑ Bougeard G, Hadj-Rabia S, Faivre L, Sarafan-Vasseur N, Frebourg T. The Rapp-Hodgkin syndrome results from mutations of the TP63 gene. Eur J Hum Genet. 2003 Sep;11(9):700-4. PMID:12939657 doi:http://dx.doi.org/10.1038/sj.ejhg.5201004
- ↑ Kantaputra PN, Hamada T, Kumchai T, McGrath JA. Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectodermal dysplasia. J Dent Res. 2003 Jun;82(6):433-7. PMID:12766194
- ↑ Bertola DR, Kim CA, Albano LM, Scheffer H, Meijer R, van Bokhoven H. Molecular evidence that AEC syndrome and Rapp-Hodgkin syndrome are variable expression of a single genetic disorder. Clin Genet. 2004 Jul;66(1):79-80. PMID:15200513 doi:10.1111/j.0009-9163.2004.00278.x
- ↑ Leoyklang P, Siriwan P, Shotelersuk V. A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet. 2006 Jun;43(6):e28. PMID:16740912 doi:43/6/e28
- ↑ Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998 Sep;2(3):305-16. PMID:9774969
- ↑ Sasaki Y, Ishida S, Morimoto I, Yamashita T, Kojima T, Kihara C, Tanaka T, Imai K, Nakamura Y, Tokino T. The p53 family member genes are involved in the Notch signal pathway. J Biol Chem. 2002 Jan 4;277(1):719-24. Epub 2001 Oct 18. PMID:11641404 doi:10.1074/jbc.M108080200
- ↑ Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, Darimont BD, Duijf PH, Van Bokhoven H, McKeon F, Dotsch V. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol. 2002 Dec;22(24):8601-11. PMID:12446779
- ↑ Ghioni P, Bolognese F, Duijf PH, Van Bokhoven H, Mantovani R, Guerrini L. Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol. 2002 Dec;22(24):8659-68. PMID:12446784
- ↑ Zeng SX, Dai MS, Keller DM, Lu H. SSRP1 functions as a co-activator of the transcriptional activator p63. EMBO J. 2002 Oct 15;21(20):5487-97. PMID:12374749
- ↑ Heyne K, Willnecker V, Schneider J, Conrad M, Raulf N, Schule R, Roemer K. NIR, an inhibitor of histone acetyltransferases, regulates transcription factor TAp63 and is controlled by the cell cycle. Nucleic Acids Res. 2010 Jun;38(10):3159-71. doi: 10.1093/nar/gkq016. Epub 2010, Jan 31. PMID:20123734 doi:10.1093/nar/gkq016
- ↑ Mitchell K, O'Sullivan J, Missero C, Blair E, Richardson R, Anderson B, Antonini D, Murray JC, Shanske AL, Schutte BC, Romano RA, Sinha S, Bhaskar SS, Black GC, Dixon J, Dixon MJ. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet. 2012 Jan 13;90(1):69-75. doi: 10.1016/j.ajhg.2011.11.013. Epub, 2011 Dec 22. PMID:22197488 doi:10.1016/j.ajhg.2011.11.013
|