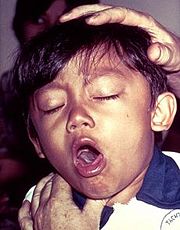
A young boy coughing due to pertussis.
Introduction
Pertussis Toxins (PT) is a protein-based exotoxin and major virulence factor produced by the bacterium Bordetella pertussis.[1] PT causes pertussis, which is also known at whooping cough and is highly contagious bacterial disease. The disease is caused by the bacterium colonizing the respiratory tract where it then establishes an infection.[2] This disease had been characterized by severe coughing that can last up to six weeks and in some countries lasting nearly 100 days.[2]It has been documented in some cases that PT can cause subconjunctival hemorrhages, rib fractures, hernias, fainting and vertebral artery dissection.[3]
As of 2010, the worldwide incidence of whooping cough has been estimated to 48.5 million cases and nearly 295,000 deaths per year.[4] With that in mind, whooping cough can affect people of any age; however, before vaccines were available the disease was most common in infants and young children but now children are immunized and the high percentage of cases are seen among adolescents.
Structure
The pertussis toxin has been characterized as being an AB toxin meaning that there are 2 subunits: A subunit possesses the enzyme activity and the B subunit possesses the receptor binding portion. PT in particular is an AB5 toxin consisting of a six-component protein complex, and the multiple subunits of the complex are not identical in composition.[1] With that in mind, this protein is a hexamer containing a catalytic (S1) subunit that is tightly associated with the pentameric cell-binding component (B-oligomer).[1] The S1 component is a single subunit while the B-oligomer is a pentamer composed of four types of subunits: , , two copies of , and .[1] The overall structure of PT . These subunits are encoded by ptx genes, which are encoded on a large PT operon that includes additional genes as well such as Pti genes. Together the PT and Pti proteins form the PT secretion complex and toxin itself.[5]
Pertussis Toxin activation
Pertussis Toxin by itself is harmless unless activated. From multiple studies, it has became clear that there is a direct interaction between Adenosine triphosphate (ATP) and pertussis toxin which leads to activation.[1][6]The direct effect of ATP is to destabilize the interaction between the S1 subunit and the B-oligomer by binding to the B-oligomer.[1] This interaction relaxes the toxin by facilitating the subsequent reduction of a disulphide bond in the S1 subunit.
The main interaction that leads to the destabilization is the favorable hydrogen bonding and electrostatic interaction between the triphosphate moiety and five positively charged amino acids:. In contrast, the negatively charged carboxyl terminus of subunit S1 interacts unfavorably with the negative charges of the triphosphate moiety, causing a displacement of the C-terminal of therefore, the repulsion between the triphosphate moiety and the C terminus of subunit S1 forms the mechanism by which the interaction between S1 and the B-Oligomer is destabilized.[1]The details of the protein-ATP interactions can also be seen here.[1]
Mechanism of pathogenesis
The disease pertussis has two stages. The first stage is colonization of the upper respiratory tract where B. pertussis adheres by using filamentous hemagglutinin (FHA) and cell bound pertussis toxin PTx. This can be visualized in the following image of the colonization of tracheal epithelial cells by Bordetella pertussis .
[7]
The second stage is the toxemic stage which follows the colonization stage. The PT B subunit oligomer uses S2 and S3 as adhesins, and they bind the bacteria to host cell receptors. S2 and S3 utilize different receptors on host cells. S2 binds specifically to a glycolipid, which is found primarily on the ciliated epithelial cells. S3 binds to a glycoprotein found mainly on phagocytic cells. After binding, the toxin is taken up by an endosome and transported from the plasma membrane via the Golgi apparatus to the endoplasmic reticulum (ER) where finally membrane translocation occurs.[8] The destabilization of PT occurs in the ER prior to membrane translocation. After binding of ATP, cleavage of the single disulphide bond by protein disulphide isomerase (PDI) occurs in subunit S1 and is believed to trigger a conformational change necessary to expose the active site to its substrates.[1] The reducation step takes place after interaction of PT with ATP.
After destabilization, the S1 becomes active and catalyzes the ADP-ribosylation of the alpa-subunit of regulatory trimeric G-proteins (Giα) host protein.[1][6] This then prevents Giα from inhibiting adenylate cyclase and leads to an increase in intracellular levels of Cyclic adenosine monophosphate (cAMP). The conversion of ATP to cyclic AMP cannot be stopped and intracellular levels of cAMP increase.[9]This has the effect to disrupt cellular function/signaling, and in the case of phagocytes, to decrease their phagocytic activities such as chemotaxis, engulfment, the oxidative burst, and bacteridcidal killing.[10]
Treatment
Early treatment of pertussis is the only effective way to treat the bacterial infection. If treatment for pertussis is started early in the course of illness during the 1 to 2 weeks before severe coughing occurs, the symptoms may be lessened.[11]The preferred antibiotics are that inhibit protein synthesis such as: Erythromycin, Clarithromycin, and Azithromycin.[12] If the diagnosed to late, antibiotics will not alter the course of the illness since the bacteria is already producing the PT toxin.
The primary method of prevention for pertussis is vaccination. The DTaP vaccine is used and this vaccine is composed of diphtheria, tetanus, and pertussis. The pertussis component is acellular; this acellular component are selected antigens (inactivated pertussis toxin (toxoid) and filamentous hemagglutinin) of pertussis that induces adaptive immunity.[13]
Conclusion
The findings of PT activation was significant since it illustrated a better understanding for the pathogenesis of the toxin. The key features of this proposal is that ATP binding signals the arrival of the PT in the endoplasmic reticulum by acting as a molecular sensor.[1] This detection of the PT in the ER at the same time triggers dissociation of the holotoxin prior to membrane translocation.[1] Therefore, the dissociation is due to ATP binding destabilization, reduction by protein disulphide isomerase, and proteolytic cleavage.[1]


{kind=link}
{kind=link}
{kind=link}