Functional Overview
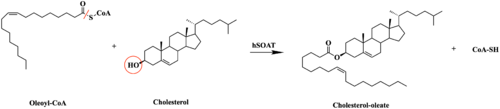
Figure 1. Esterification reaction of oleoyl-CoA and cholesterol catalyzed by SOAT. This reaction results in the formation of cholesteryl esters and CoA-SH byproduct.
Sterol O-acyltransferase(SOAT), otherwise known as Acyl-coenzyme A:cholesterol acyltransferase(ACAT), is the first discovered member of the membrane-bound O-acyl
transferase or MBOAT enzyme group. MBOAT enzymes are responsible for the transfer of
acyl chains onto multiple types of substrates within the cell. There are 11 MBOAT enzyme types that can be found in humans, all of which serve a different function in the overall makeup of human biology.
[1]
SOAT specifically catalyzes the esterification of cholesterol for efficient storage within the cell (Figure 1). As a membrane lipid, cholesterol is responsible for controlling the fluidity and integrity of the membrane, along with other important biological processes. When there are high concentrations of cholesterol in the cell, cholesteryl esters can be formed for storage within the membrane.[1]
Structure
Tertiary Structure
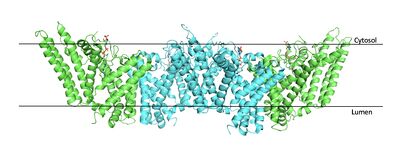
Figure 2. Tetramer unit of SOAT shown in position within the membrane. The dimer units are identical, as indicated by the corresponding green and blue regions.
PBD 6P2P The biological assembly of SOAT is a or a (Figure 2). Functionally, the units of SOAT are identical and are stabilized by hydrophobic
van der Waals interactions between residues at the . Mutating these residues inhibits enzyme activity, suggesting that the dimer unit of SOAT is critical for enzyme function.
[1] Each dimer consists of two identical units, individually made up of nine labeled TM1 through TM9 (Figure 3).
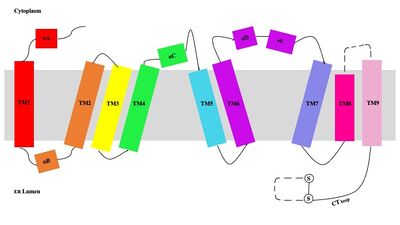
Figure 3. Labeled helices of SOAT within the membrane. The van der Waals interactions at the dimer interface stabilize the dimer between the TM1 helix of one monomer unit and the TM6
lumenal segment and TM9
cytosolic segment of the other monomer unit. Essential helices (TM1, TM5, TM6, and TM9) from the two monomers form the entrance tunnels and catalytic active site.
[2]
Tunnel System
An important structural element of SOAT is the tunnel system through which substrates enter and exit.
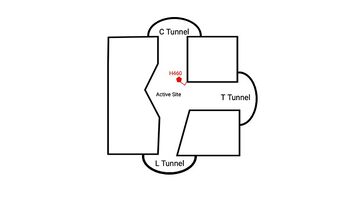
Figure 4. 2D layout of the proposed SOAT tunnel system. The C tunnel opens into the cytosol and the L tunnel opens to the lumen. The T tunnel opens into the membrane, but is not quite oriented at a 90 degree angle as depicted here.
There are three main tunnels in each monomer: the cytosolic (C) tunnel opens to the cytosol, the transmembrane (T) tunnel opens to the membrane, and the lumenal (L) tunnel opens to the lumen (Figure 4).
[2] The is the entrance site for the acyl-CoA substrates, allowing them acess to the active site. Residues exhibit hydrogen bonding interactions with polar atoms of coenzyme A to help stabilize the substrate within the binding pocket. Surface representations of SOAT indicate that there are two alpha helices that block the entrance to the C tunnel, therefore a conformational change needs to occur before the substrate can enter the tunnel. The opens into the membrane and provides access for cholesterol to enter the active site. At the active site, SOAT catalizes the esterification reaction and the products exit through the tunnels. The CoA-SH product exits through the C tunnel and is released back into the cytosol. The cholesteryl ester product exits through either the T tunnel into the membrane or through the
into the lumen of the cell. [2]
Active Site
Within the binding pocket, there are several . Their high preservation suggests that the local environment of the binding pocket plays a major role in SOAT activity, but their specific interactions are currently not well studied. However, have been identified as key catalytic residues.[1] Histidine, commonly used as the catalytic base to initiate acyl transferase reactions, is assumed to be the most important catalytic residue in SOAT.[3] This was confirmed as mutating H460 to alanine completely abolished enzymatic activity.[4] Additionally, H460 is highly conserved across a variety of species, further emphasizing its importance in SOAT catalysis.[1] It is hypothesized that N421 stabalizes the transition state via hydrogen bonding with coenzyme A.[2] Additionally, mutations of W420 to alanine render SOAT nonfunctional, indicating that it must be essential for catalytic activity. However, its role in the mechanism is not explicitly hypothesized. We believe that it plays a role in substrate binding through with the acyl chain of coenzyme A.
Catalytic Mechanism
The substrate of interest, , is shown bound to SOAT to visualize the binding pocket. It must be noted that cholesterol, the other substrate involved, was never correctly imaged in the active site of SOAT. Upon binding of oleoyl-CoA and cholesterol to the SOAT , the distal-most nitrogen on H460 acts as a base catalyst to deprotonate the hydroxyl group of cholesterol. This leaves the cholesterol oxygen with a negative charge, making it a good
nucleophile. The nucleophilic oxygen then attacks oleoyl-CoA at its carbonyl carbon, kicking electron density up to the carbonyl oxygen. The transition state is stabilized by and the newly protonated H460 (Figure 5).
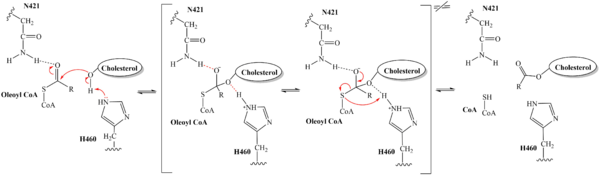
Figure 5. Mechanism for the esterification reaction of SOAT with arrow pushing. From the transition state, excess electron density on the carbonyl oxygen is collapsed back into a double bond. This causes the bond between the carbonyl carbon and sulfur to break, shifting electron density to the sulfur atom. To complete the mechanism, the negatively charged sulfur would reclaim the hydrogen from protonated H460 (Figure 5). CoA-SH would exit the active site as a leaving group, leaving its oleoyl chain attached to cholesterol in the form of a cholesteryl ester.
It should be noted that this mechanism is largely hypothesized based on our understanding of esterification reactions and the residues involved. Further analysis is needed to confirm the proposed steps.
Inhibitors
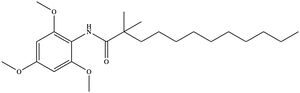
Figure 6. Structure of the CI-976 inhibitor. Depending on concentration and exposure, SOAT activity can be inhibited by CI-976 (Figure 6). When exposed, CI-976 locks itself in the of the enzyme. The becomes sandwiched between the catalytic residues W420, N421, and H460. The interaction with these residues, as well as the location of the trimethoxyphenyl head, indicate that CI-976 acts as a
competitive inhibitor of SOAT by preventing substrates from accessing the catalytic center. Similar to interactions with the substrates, mutating the key catalytic residues (N421A, H460A, and H460N) results in a smaller effect of the inhibitor on the thermostability of the enzyme.
[1]
Biological Relevance
SOAT has gained interest when looking at its biological relevance because it has the ability to use a wide range of sterols in it’s mechanistic activity. The wide variety of substrates has led to SOAT being focused on as a potential drug target for many different diseases. Alzheimer’s disease, atherosclerosis, and several types of cancers have show success in treatments when targeting the SOAT enzyme’s catalytic mechanism. [1] In summary, targeting SOAT could be an effective means for treating various diseases. Aberrant quantities of cholesteryl esters seem to hinder various cellular processes; thus, inhibiting SOAT expression and functionality could help reduce these adverse effects. Overall, SOAT plays an important role in cholesterol homeostasis and future research of this enzyme could lead to the discovery of therapeutic treatments for different illnesses.
Alzheimer's Disease
Alzheimer’s disease (AD) is a progressive disease that severely hinders a person’s memory and other cognitive functions. AD is the result of a significant increase in beta-amyloid (Aβ) peptide concentration. [5] Previous studies have found that the amount and distribution of intracellular cholesterol plays an important role in regulating Aβ production.[6] Therefore, SOAT inhibition could be an effective therapy for treating AD because it would reduce cholesteryl ester formation in the brain and help lower Aβ generation as well. [5] [6]
Atherosclerosis
Another disease SOAT inhibition could help treat is atherosclerosis. Buildup of cholesteryl esters from SOAT catalysis has been shown to be partially responsible for foam cell formation, one of the major indicators of atherosclerosis. Consequently, SOAT inhibitors have been studied as potential drug targets for this disease.[7]
Cancer
Increased expression of SOAT and abnormal accumulation of cholesteryl esters has also been found in multiple cancers including ovarian cancer. Therefore, inhibiting SOAT and exhausting cholesteryl ester concentrations has shown to have anti-tumor effects in terms of monitoring apoptosis, cell proliferation, and migration and invasion properties. Therapies that target SOAT regulation and expression levels could thus lead to potential treatments for ovarian and other types of cancer.[8]






