Introduction
Three different isoforms of VDAC have been identified, VDAC1, VDAC2, and VDAC3. VDAC1 has been best studied, whereas only limited information regarding the cellular functions of VDAC2 and VDAC3 is available .VDAC1 is a protein present in the outer mitochondrial membrane(OMM) and in other cellular compartments such as the plasma membrane.VDAC1 controls the metabolic and energy cross-talk between mitochondria and the rest of the cell, mediating the fluxes of ions, nucleotides, and other metabolites across the OMM . This protein is involved in several cellular processes.
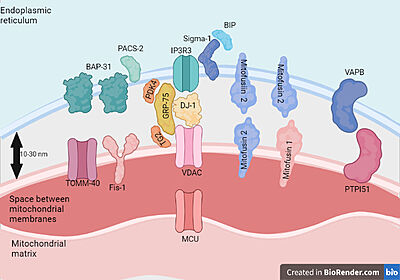
Proteins present in mitochondria and endoplasmic reticulum membranes that interact with each other.Feito com Biorender.com
VDAC1 has been shown to be a regulator of OMM permeability to Ca2+.Mitochondria play a major role in different events beyond theircritical bioenergetics function of supplying ATP, such as in cellsignaling events, inter-organellar communication, aging, cell pro-liferation, disease, and apoptosis. Apart from their metabolic role,mitochondria are also a major hub of cellular Ca2+homeostasis thatis fundamental for a wide range of cellular activities, such as controlof oxidative phosphorylation, modulation of cytosolic Ca2+signals,cell death, secretion, and the production of reactive oxygen species(ROS)
Relevance
As VDAC1 is a mitochondrial membrane protein, it is involved in cell metabolism in addition to mitochondrial-dependent apoptosis processes and regulates calcium homeostasis and oxidative stress.VDAC1 is highly Ca2+-permeable and modulates Ca2+ access to the mitochondrial intermembrane space. Intramitochondrial Ca2+ controls energy metabolism by enhancing the rate of NADH production via modulating critical enzymes in the tricarboxylic acid cycle and fatty acid oxidation. Mitochondrial [Ca2+] is regarded as an important determinant of cell sensitivity to apoptotic stimuli and was proposed to act as a “priming signal,” sensitizing the organelle and promoting the release of pro-apoptotic proteins.Intracellular Ca2+concentration ([Ca2+]i) regulates a number of cellular and intercellular events, such as the cell cycle, proliferation, gene transcription, and cell death pathways, as well as processes like muscle contractility and neuronal processing and transmission. The alteration of Ca2+ homeostasis is closely related with various cancer hallmarks, including proliferation, migration, angiogenesis, invasion abilities, and resistance to cell death.VDAC1 has also been recognized as a key protein in mitochondria-mediated apo-ptosis, contributing to the release of apoptotic proteins located in the inter-membranal space (IMS) andregulating apoptosis via association with pro- and anti-apoptotic members of the Bcl-2 family of pro-teins and hexokinase.
Structural highlights
Human VDAC1 (hVDAC1) adopts a β-barrel architecture composed of 19 with strands β1 and β19 being in parallel connformation and an located horizontally midway within the pore. N-terminal region of VDAC1, consisting of 25 amino acids,lies inside the channel pore and possesses different degrees of α-helical content in each of the three proposed structures.Various studies using purified rat liver , brain mitochondria or recombinant human VDAC1 have reported that both soluble purified and membrane-embedded VDAC1 can assemble into , trimers, tetramers and higher oligomeric states. VDAC1 oligomerization was also demonstrated in VDAC1-reconstituted liposomes.VDAC1 oligomerization involves several different interactions, such as hydrophobic interactions between β‐strands, hydrophilic interactions between loop regions and one of the β‐strands, and protein–lipid interactions.
Return to or structure
Neurodegenerative disease
![Alterations in VDAC1 observed in ALS[2].](/wiki/images/thumb/e/e1/ALS_dysfunctions.png/500px-ALS_dysfunctions.png)
Alterations in VDAC1 observed in ALS[2].
Several studies have already observed the participation of VDAC1 in mitochondrial dysfunctions observed in several neurodegenerative diseases and constitutes a point of accumulation of protein aggregates with Tau,β amyloid,SOD1 that are present in these pathologies. Due to the involvement of VDAC1 in metabolism and processes of apoptosis this protein becomes a possible therapeutic target of these diseases.The Cu/Zn superoxide dismutase (SOD1) associates with about 20% of familial ALS (fALS) cases and over 180 mutant forms of enzymatically active or inactive SOD1 have been characterized in humans (http://alsod.iop.kcl.ac.uk). In affected tissues, toxic effects of SOD1 mutants are related to the formation of misfolded SOD1 aggregates upon the mitochondrial surface, leading to morphological degeneration and malfunctioning of the organelle.In the spinal cord from ALS patients, voltage dependent anion selective channel isoform 1 (VDAC1) represents the docking site on the outer mitochondrial membrane for ALS-linked SOD1 mutants.In ALS, the VDAC1-SOD1 mutant interaction strongly affects the functional properties of VDAC1
channel suggesting a role in the impairment of the bioenergetics metabolism and oxidative stress of
ALS motor neurons.It is also known that low levels of hexokinase I (HK1) in the spinal cord make motor neurons more susceptible to ALS in comparison to other tissues. In particular, this reduction of HK1 levels increases the availability of VDAC1 to interact with mutant SOD1, thereby facilitating mitochondrial dysfunction and cell death.he abnormal accumulation of misfolded proteins and dysfunctional mitochondria is a distinctive feature of ALS and many neurodegenerative diseases.VDAC1 is the receptor of HK1 and of many other enzymes and metabolites (not shown), but not of SOD1WT Conversely, in ALS, SOD1G93A is
able to bind deamidated VDAC1 impairing thus the binding of HK1 and likely of others physiological interactors A is able to bind deamidated VDAC1 impairing thus the binding of HK1 and likely of others physiological interactors. We propose that deamidation of VDAC1 residues parallels oxidative stress levels, leading to the enhancement of SOD1 mutant binding to VDAC1. As a consequence, VDAC1 channel conductance and metabolic exchanges through VDAC1 are gradually affected, promoting a growing mitochondrial dysfunction. The oxidative damage that accumulates with disease
progression produces therefore dysfunctional mitochondria tagged with deamidated VDAC1 that could be selected and forwarded to mitophagy or fission processes.

![Alterations in VDAC1 observed in ALS[2].](/wiki/index.php/Image:ALS_dysfunctions.png)
