Introduction
Overview
Stearoyl-CoA Desaturase-1 is an integral membrane protein located in the endoplasmic reticulum and is conserved across all eukaryotes [1]. The SCD-1 pictured is expressed in Mus musculus (house mouse). The human homolog, SCD1, shares 85% sequence identity with all four SCD’s found in M. musculus (SCD1-SCD4). The expression of SCD1 is seen mainly in the liver and brain [2].
SCD is an enzyme which catalyzes desaturation of a double bond within a saturated fatty acid chain (Figure 1). The addition of a double bond is necessary for the biosynthesis of monounsaturated fatty acids such as: cholesterol, phospholipids, and triglycerides. The enzyme’s main function is
lipid biosynthesis as well as regulating gene expression for
lipogenesis [1]. SCD1 is regulated by transcription and its promoter has multiple binding sites for transcription factors that assist with regulation of lipogenesis
[2]. It was discovered that when mice were SCD1-deficient, there was no obesity seen in the mice
[1] This is why SCD1 is a popular target in treating metabolic diseases. Functioning SCD1 creates the balance between the accumulation and use of fats in the body.
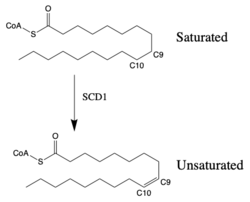
Figure 1: Saturated vs. Unsaturated Fatty Acid Chain. SCD-1 is interacts with either of the two different substrates: stearoyl-CoA or palmitoyl-CoA. When SCD1 interacts with stearoyl-CoA and performs desaturation, the product is oleoyl-CoAand has the first cis-double bond introduced into the fatty acid chain. The introduction of the cis-double bond into the hydrocarbon chain will increase fluidity of the lipid bilayer. The process of desaturation is tightly regulated by multiple transcription factors.
The desaturase enzyme also works in combat with inhibitors. SCD1 is affected by hormones, growth factors, and nutritional status [2]. Leptin is a hormone that plays a role in regulation of energy homeostasis and is also able to stop SCD-1 expression by activating specific transcription factors to bind to SCD1 promoter and overpower the insulin signals [2]. Other negative regulators of SCD include estrogen and glucagon. SCD1 can also be inhibited by one’s nutritional status because of the production of polyunsaturated fatty acids (PUFAs), causing a high concentration of fats in the body.
Structure
SCD1 has arranged in a “mushroom"-like shape [3]. The cytosolic domain makes up the crown of the enzyme and the TM domains make up the stem. There is a conserved and catalytic that coordinates two metal molecules (iron or zinc), and make up the di-metal center within the binding pocket.
Binding Pocket
SCD1 consists of a hydrophobic deep within the protein that the substrates will enter [4]. The tunnel is regioselective and stereospecific such that the substrate’s binding site lines up C9 and C10 at the kink of the V-shaped tunnel with the di-iron center that consists of an oxygen molecule bound to one of the metals. Precise placement of the atoms near the two iron metals provides the tunnel with regioselectivity and stereospecificity, stabilizing the substrate to allow for desaturation at carbons 9 and 10. The kink is formed by two conserved [1]. It is at this kink of the tunnel where desaturation occurs.
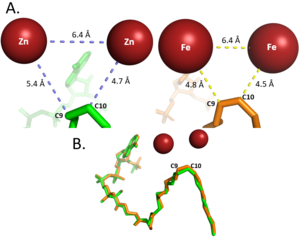
Figure 2: Differences between zinc and iron di-metal interactions with acyl-CoA. Metal Cations
Typically, SCD1 contains a in the core of the protein that contributes to its desaturation function. Crystallized structures of SCD1 indicated a substitution of zinc as the di-metal center instead of iron[1]. It is proposed that the difference in size of zinc and iron (8.8 Å and 9.2 Å respectively) would not affect the structure of SCD. However, the substitution of zinc as the di-iron center is found to inhibit the function of SCD1 due to a farther distance between the two metals compared to iron (Figure 2)[1].
In other di-metal centered enzymes such as ACP desaturase and ribonucleotide reductase, the enzymatic mechanism involves an oxo-bridge: a water molecule that is recruited by the di-iron center to be directly involved in the desaturase mechanism. This water gets deprotonated by the two metal ions and it becomes nucleophilic enough to attack the substrate. Based on electron density mapping of SCD1, this oxo-bridge formation is suggested to be short-lived [3].
Mutations and Substrate Specificity
Different desaturases have varying catalytic rates depending on the carbon chain length of the substrate. For example, desaturase ChDes1 is found in arctic copepod Calanus hyperboreus and contains a threonine instead of a at position 104 in SCD1 and other desaturases which causes the loss of desaturase function in 26C substrates while retaining activity with 18C substrates. In SCD3, stacked mutations I112A and Q113L changed the enzyme’s specificity from a 16C to a 18C desaturase [1]. It is hypothesized that mutations that occur on TM2 near the substrate binding tunnel change the substrate specificity of SCD1.
Function
Palmitoyl and Stearoyl CoA are substrates of SCD1. These substrates enter the hydrophobic V-shaped tunnel inside SCD1. Hydrogens are removed at the C9, then C10 to introduce the double bond through a desaturation mechanism.
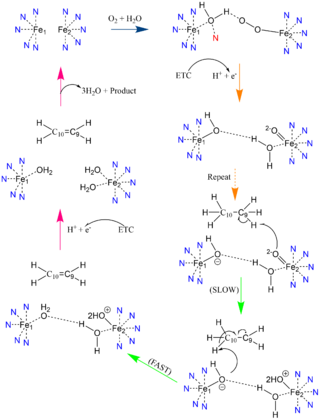
Figure 3: Proposed mechanism of SCD1 based off Shen et al. 2020 [3]. Nitrogens colored blue represent the histidine stabilization of the di-iron complex while the red nitrogen is the water-stabilizing asparagine.
Desaturation Mechanism
Due to the novel coordination of histidine residues and lack of presence of a stable oxo-bridge, the mechanism of desaturase activity for SCD1 is unknown. For typical desaturases, the elements involved in catalysis include the di-metal center, metal-bound water and oxygen, electrons from an Electron Transport Chain (ETC), and the acyl-CoA substrate[3]. For the proposed mechanism, catalysis occurs in four phases: the binding of water and oxygen to the di-metal complex using Asn261 (blue arrow), the nucleophilic activation of the water/oxygen using the ETC (orange arrows), the interaction of the complex with the substrate (green arrows), and the recovery of the enzyme (pink arrows) (Figure 3). The rate-limiting step of the mechanism is the primary elimination reaction on the substrate due to the nucleophilic attack occurring on an already stable C9-C10 sigma bond.
Product Conversion and Release
The acyl-CoA substrate, due to it containing all sigma bonds, is able to freely rotate around its carbon axes to fit into the deep, kinked binding pocket. Upon double bond formation between C9 and C10, the substrate rotates from conformation across C9 and C10.The double bond also restricts rotation along carbons 9 and 10 which likely prevents the substrate from exiting through the site that it entered[1]. To allow for , it is hypothesized that a hydrogen bond between Gln143 and Thr257 is broken which creates a hole below the kink into the hydrophobic core of the enzyme. This would allow for the lateral transfer of the product out of the binding pocket without removing the double bond.
Related Disease
Diabetes and Obesity
Metabolic-related issues such as obesity and diabetes are linked to having a diet high in saturated fats and simple sugars [5]. SCD1 catalyzes the desaturation of fully saturated fatty to monounsaturated fatty acid chains and acts as the rate limiting step for this type of desaturation mechanism [6]. The deficiency and/or inhibition of SCD1 results in increased insulin sensitivity, reduced body obesity and resistance to diet-induced obesity.
In the medical field, SCD1 is being used as a target for treatment of obesity, diabetes, and other metabolic diseases [1]. In previous research, a decrease in SCD1 has shown to decrease the expression of lipogenic genes such as fatty acid synthase (FAS), acetyl CoA carboxylase(ACC), and cholesterol 7 alpha-hydroxylase (CYP7A 1) [5]. SCD1-deficiency causing lowered expression of these fat-synthesizing genes also represses glycolysis and gluconeogenesis. SCD1 deficiency shows an increase in insulin sensitivity and increase resistance to diet-influenced obesity [7]. Changes in cellular concentration of this enzyme can cause positive or negative affects to metabolic processes that are linked with diabetes and obesity.
Cancer
SCD1 works to keep a balance between saturated and monosaturated fatty acids, and in turn, this helps prevent the build up of toxic saturated fats inside the cell[8]. Increased expression levels of SCD1 has shown to enhance cancer cell proliferation. The concentration of monounsaturated fatty acids (MUFAs) is much higher in a cancer cell versus a healthy cell. When expression of SCD1 in breast cancer patients was examined, results indicated that increased expression of SCD1 was associated with shorter patient survival [8]. This is most likely due to the promoter region of SCD1. A transcription factor named SREBP-1 binds tightly to the promotor of SCD1, and both become upregulated when the PI3K signaling pathway is active[8]. PI3K/Akt signalingis involved with cancer cell proliferation when the pathway becomes disrupted [9].
Inhibition of SCD1 can stop cancer cell propagation both directly and indirectly [2]. The enzyme directly regulates signaling pathways, and indirectly interrupts the ratio of monounsaturated to saturated fatty acids. SCD1 is proving as a positive potential anticancer therapy, considering the difficulty many patients face when going through multiple rounds of chemotherapy. The reason cancer cells often are able to become resistant to chemotherapeutic drugs is because they possess stem cell-like phenotypes [2]. SCD1 inhibition has shown to repress tumor growth from cancer stem cells (CSC) and prevent any new proliferation of the CSC [10].



