Introduction
is the serine protease that catalyzes the penultimate step in blood coagulation. It is activated from its zymogen, prothrombin, at the site of tissue injury by Factor Xa (FXa) and its cofactor FVa in the presence of phospholipid membrane and calcium. Thrombin is then able to catalyze the cleavage of fibrinogen to insoluable fibrin which spontaneously polymerizes to form a stable clot.[1][2] Thrombin also acts as a procoagulant by:
Activity of thrombin is regulated physiologically by the serpin inhibitors:
Generation of thrombin is decreased when thrombin reaches endothelial lining and interacts with thrombomodulin which significantly increases activity of thrombin in activating protein C (PC -> APC).[9] This enzyme will go on to inactivate FVa[10][11] and FVIIIa[11] , cofactors for activation of prothrombin[12] and FXa[13], respectively.
By balancing substrate specificity, activity, and inhibition thrombin plays a central role in the blood coagulation cascade. [4]
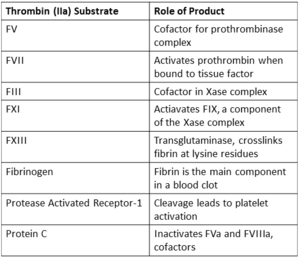
Coagulation related substrates of thrombin, excluding serpin inhibitors.
The Thrombin Life Cycle
Upon tissue damage tissue factor (TF) is released by subendothelial cells. This interacts with circulating FVIIa, a zymogen-like serine protease, significantly increasing its activity. The FVIIa-TF complex (extrinsic Xase) activates FVII, FIX, and FX. The now active FXa cleaves prothrombin bound to membranes through its gamma-carboxyglutamyl (Gla) domain, activating it to thrombin.
Thrombin interacts with platelet membrane protein GpIbα and subsequently cleaves protease activated receptor-1 (PAR1) causing a signaling cascade which leads to platelet α-granule release and membrane flipping exposing the negatively charged phosphatidylserine. The platelet alpha granules contain the physiologically relevant pool of FVa. [14]
Thrombin also causes activation of FIX, through FXI cleavage, and FVIII which form the Xase complex to activate FX. FVa and FXa form the prothrombinase complex in the presence of calcium and phospholipid. It causes rapid activation of prothrombin to thrombin. This increase in thrombin allows sufficient fibrinogen to be cleaved to fibrin which is able to polymerize to form a stable blood clot.
Further supporting coagulation, thrombin activates FXIII, a transglutaminase that crosslinks fibrin at lysine residues.
The structure of thrombin facilitates its inactivation. Once the endothelial lining is reached thrombin binds heparin or related glycosaminoglycans. This facilitates its inactivation by serpin inhibitors antithrombin and heparin cofactor II. In addition, thrombin will interact with thrombomodulin which significantly increases its catalytic efficiency activating protein C. Activated protein C inactivates FVIIa and FVa thus down regulating thrombin generation.
Thrombin plays a critical dual role in blood coagulation. It must be both promiscuous and specific so that it may complete both procoagulant and anticoagulant functions. The structure of thrombin facilitates its important physiologic functions.
Prothrombin Activation
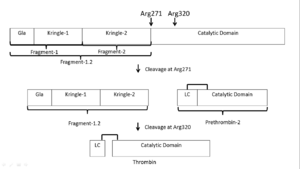
Activation scheme of in vivo activation of prothrombin by FXa in the absence of FVa.
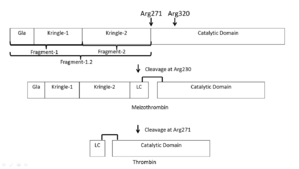
Activation scheme of in vivo activation of prothrombin by the prothrombinase complex in presence of calcium and a phospholipid bilayer
Prothrombin is the zymogen form of thrombin. From N-terminal to C-terminal it consists of a Gla domain, two kringle domains, and a catalytic domain. The Gla domain is formed by vitamin K dependent carboxylation of glutamate residues.[15]
Prothrombin is activated by prothrombinase which consists of FXa, FVa, calcium, and a phospholipid surface. In vivo the first cleavage occurs at the R320-I321 bond, corresponding to residues 15-16 in chymotrypsin which is the N-terminus of the B chain, producing meizothrombin.[16] Subsequent cleavage at R271-T272 yields thrombin.[16] The initial cleavage can also occur at R271 resulting in prethrombin-2 which will then be cleaved at R320 to produce thrombin.[17]
After cleavage by prothrombinase the new B chain N-terminus (Ile16) folds into the core protease domain and forms a salt bridge with Asp194.[16] This leads to stabilization of regions of the 180s-loop, Na+ binding loop, and γ-loop (zymogen activation domains). These changes provide the correct conformation for the S1 pocket and oxyanion hole for catalysis.[16][18]
Structure and Function
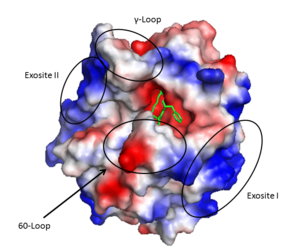
Thrombin (1PPB) overlayed with electrostatic surface. Structural features 60-loop, γ-loop, exosite I, and exosite II labeled
Thrombin is a α/β heterodimer composed of a 36 amino acid A chain and 259 amino acid B chain connected by a bridge between Cys1 and Cys122, in addition to 3 other intrachain disulfide bonds.[19] Its overall fold is similar to trypsin and chymotrypsin and it belongs to the peptidase S1 protease family[20]. It is an overall spherical protein with approximate dimensions of 45 Å X 45 Å X 50 Å.[19]
Important structural features include:
- A prominent active site cleft flanked by the 60- and γ-loops
- A sodium binding loop
- Two surface patches referred to as exosite I and exosite II.
The is mostly helical and is wound around the B chain and shaped like a boomerang. It is bound to the B chain mostly through side chain interactions including a salt bridge and a hydrogen bond cluster at residues D14, E8, and E14c.[19] Furthermore the C-terminus region forms a short amphipathic helix with hydrophobic side chains interacting with the B chain.[19]
The contains the active site of the protein and has numerous notable structural features. The active site is formed at the rims of two interacting 6 stranded (N-terminal barrel in red and C-terminal barrel in orange) which are surrounded by 4 helical regions and many turns.
The serine protease (wiki) residues, based on chymotrypsin numbering, are Ser195, His57, and Asp102. As is common with serine proteases, an hole is formed by backbone amides of Ser195 and Gly193.[16] This has the functional role of stabilizing the oxyanion intermediate involved in the serine protease mechanism by hydrogen bonding to the oxygen of the P1 residue (standard substrate-protease nomeclature [21]. In addition, since thrombin cleaves after Arg/Lys the , formed by the 180s- and 220s- loops, has Asp189 at the base to form a salt bridge with the incoming substrate. Furthermore, the S4 binding pocket accommodates hydrophobic substrate residues.
The cleft rims are formed by the hydrophobic and rigid (residues L60, Y60a, P60b, P60c, W60d, D60e, K60f, N60g, F60h, T60i, and N60g) and the (residues T147, W147a, T147b, A147c, N147d, and V147f) while the base is mostly hydrophilic negatively charged amino acids. The cleft is deep compared to more promiscuous serine proteases, consequently substrates must either have a large loop that is cleaved or have favorable interactions with the insertion loops [22].
Many other loops project out of the B chain but most are rigid due to proline and tryptophan residues.
Exosite I is located on the B chain and had both basic and hydrophobic character. It is important in binding , platelet activated receptors, and .
Exosite II is also part of the B chain, and derived from numerous basic amino acids, this is the site of through the sulfate groups on the glycosaminoglycan. It is also the site of GpIbα binding on the platelet surface.
The is formed by the 180s- and 220s- loops. Na+ is bound by the backbone oxygens of Arg221a and Lys224 in addition to four water molecules in a classic octahedral geometry[23]. Through the covelent disulfide linkage between Cys220 and Cys 191 the sodium binding site is linked to Ser195 and the oxyanion hole.
Allostery
Binding of thrombin by sodium or at exosite I stabilizes a form of thrombin that improves substrate recognition.[16] This occurs due to energetic linkage between these sites to the S1 binding pocket and oxyanion hole. Rapid kinetic analysis suggests that thrombin is in a dynamic equilibrium that consists of a fast, slow, and inactive state[16]. There is question as to the physiologic relevance of the inactive state. Regardless, there will be a proportion of fast:slow thrombin and sodium binding to the fast form stabilizes that conformation. Indeed, mutation of the residues involved in sodium binding diminishes the activity of thrombin.[16] It should be restated, current data suggest that sodium binding does not induce a conformation change, rather, it stabilizes a conformation of thrombin that has greater activity.
Regulation and Inhibition
Thrombin is regulated by inhibition and down regulation of production.
Antithrombin and heparin cofactor II are serpin inhibitors of thrombin that bind to specific sequences of sugar residues within a heparin chain on the endothelial lining.[24] Thrombin also nonspecific to sequence through its exosite II.[8] Thus, heparin acts as a surface that is outside of the procoagulant environment of the blood clot for thrombin interaction with inhibitors.
also binds to heparin (through an EGF-like domain) and thombin, it causes a conformation in thrombin that increases activity for TAFI and protein C by 1000-fold.[25] APC then inactivates FVa and FVIIIa effectively decreasing the concentration of prothrombinase and Xase respectively, and thereby down regulating thrombin production.
Central to both of these regulation pathways are specific cofactor binding at thrombins exosites; thrombomodulin and antithrombin at exosite I and heparin at exosite II. Therefore the structure of thrombin uniquely provides it the functional properties necessary for regulation.
Secondary Structure Features
Thrombin has a in a loop containing proline 37. The dihedral (C'-Cα-N-C') angle omega is -69.4 degrees.
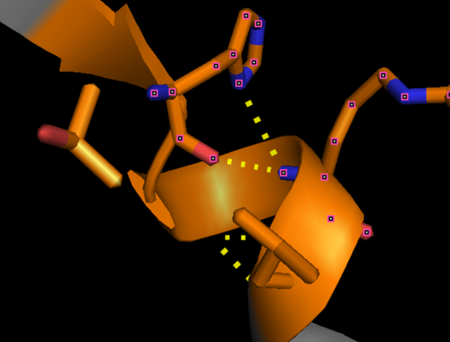
“Capping box” motif in alpha thrombin (PDB: 1PPB) represented by His230 (Ncap) side chain and main chain hydrogen bonded with the backbone nitrogen of Arg233 (N3). An additional feature is a weak hydrophobic interaction between Thr229 (N’) and Val234 (N4) termed the “hydrophobic staple.” This motif derives it’s name from the box shaped hydrogen bonding pattern.
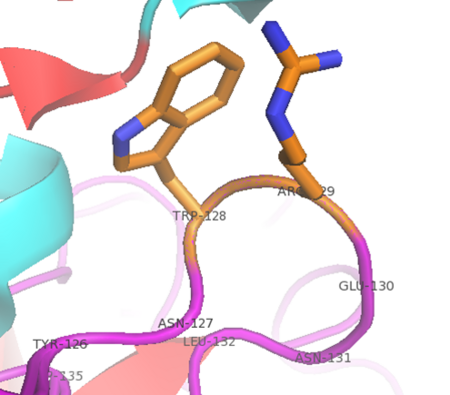
A cation-π interaction between Trp128 and Arg 129 in alpha thrombin (PDB: 2BDY). The guanidinium carbon is 3.6 angstroms from the top edge of the Trp. It is expected that the epsilon nitrogen forms the primary cation-π interaction. The electrostatic and Van der Waals interaction energies were calculated to be -3.29 kcal/mol and -3.08 kcal/mol respectively by the CaPTURE program (
http://capture.caltech.edu/result.cgi).






