Theoretical models
From Proteopedia
|
BREAKTHROUGH! In 2020, a machine learning artificial intelligence system called AlphaFold2 became able to predict the structures of a large subset of single protein chains successfully from their amino acid sequences. See CASP 14. For the AlphaFold database of predictions, and AlphaFold-based servers that will predict structure from sequence, see AlphaFold, and for practical guidance, How to predict structures with AlphaFold. |
The term theoretical model refers to a molecular model obtained using theory or artificial intelligence, such as homology modeling, energy minimization, molecular mechanics, molecular dynamics, or a machine learning system. Such theoretical models are distinguished from empirical models, which are usually obtained by X-ray crystallography, nuclear magnetic resonance (NMR), or cryo-electron microscopy.
The distinction between theoretical and empirical models is important because when theoretical models are compared with empirical models, the theoretical models often contain significant errors. In contrast, when the structure of a particular macromolecule is determined using empirical methods by different laboratories, or both by crystallography and NMR, the agreement is usually quite good.
1,390 theoretical models were historically deposited in the Protein Data Bank but removed from the main database in 2002. The structure displayed in the pages automatically generated in Proteopedia for these theoretical models should be interpreted with caution (see Category:Theoretical Model). One such database where theoretical models are allowed is the ModelArchive supported the Swiss Institute of Bioinformatics.
Contents |
Empirical Models
Empirical models are not theoretical models, but are mentioned here for the sake of completeness. Empirical models, usually determined by X-ray crystallography, nuclear magnetic resonance or cryo-electron microscopy, are the most reliable and accurate models available. Methods for judging the reliability and quality of empirical models are discussed at Quality assessment for molecular models. Independent determinations of the same protein by empirical methods generally agree within <1.0 Å root mean square deviation (RMSD) for alpha carbon atoms (reference needed).
Homology Models
Method & Limitations
Homology models, also called comparative models, are obtained by folding a query protein sequence (also called the target sequence) to fit an empirically-determined template model. The registration between residues in the query and template is determined by an amino acid sequence alignment between the query and template sequences.
- Imagine that the template’s polypeptide backbone is a folded glass tube. Now imagine that the query sequence is a thin metal chain that can be pulled through the tube. The chain (query) will adopt the same fold as the tube (template). The sequence alignment specifies how far the chain should be pulled into the tube; that is, how the residues in the query sequence match up with the structure of the template.
Errors or uncertainties in the sequence alignment result in errors or uncertainties in the homology model. Portions of the query sequence cannot be modeled reliably when there are gaps in the sequence alignment due to insertions/deletions ("indels"), or portions of the template that lack coordinates due to crystallographic disorder. Provided there is sufficient sequence identity between the query and template (at least 30%), the main chain in homology models is usually mostly correct. However, the positions of sidechain rotamers in homology models are usually unreliable.
Nevertheless, homology models are useful for seeing low-resolution features, such as which residues are on the surface or buried, which are close to other features of interest (such as a putative active site), and the overall distribution of charges and evolutionary conservation.
Attempts to improve homology models by molecular dynamics simulations have not been successful: "in most cases, simulations initiated from homology models drift away from the native structure"[1].
For further information, please see Practical Guide to Homology Modeling.
Paucity of Templates
Empirically-determined templates with adequate sequence identity are available for less than half of all protein sequences. One of the major goals of structural genomics is to increase the sequence diversity of the available empirically-determined structures that can be used as templates for homology modeling.
A number of free servers have libraries of homology models generated in advance for protein sequences, and many will create homology models for a submitted protein sequence. For more, please see
When no suitable template exists, the Structural Genomics Target Database should be searched with your sequence. In some cases, a sequence-similar protein has already been crystallized and diffracted, but the model may not have been completed, or the completed model may not yet have been deposited in the PDB. In such cases, it may be worthwhile to contact the team that has made the most progress on a closely related sequence.
See Also
- Homology modeling
- Practical Guide to Homology Modeling
- Homology modeling servers
- The list of resources at User:Wayne Decatur/Homology Modeling.
- Homology Modeling in Wikipedia
Examples
- Structure of E. coli DnaC helicase loader concerns a homology model.
Ab Initio Models
When there is no template with sufficient sequence identity to use for homology modeling, one can use ab initio or de novo folding theory, or machine learning artificial intelligence to predict the structure of a target protein sequence.
CASP
The success of structure prediction methods is assessed biannually in the Critical Assessment of techniques for protein Structure Prediction (CASP) competitions[2]. Crystallographers submit sequences which they have solved, but for which the structures have not yet been published. Modelers predict the structures which are then compared with subsequently published structures. Beginning in CASP5 (2002), the ability to predict intrinsic disorder was included[3]. Assessment of CASP results is done in a double-blind manner: the predictors do not have access to the empirical structures, and the assessors do not know the identities of the predictors, which are coded.
There have also been competitions to predict protein-protein docking interactions[4]. More recently, AlphaFold Multimer and AlphaFold 3 attempt to predict protein oligomers.
2024 CASP 16
Overall, on all fronts, AF3's modeling capabilities are at or close to the state of the art.[5] Prediction of protein oligomer complex assemblies "remains an unsolved challenge."[6] AlphaFold 2 & 3 have largely solved prediction of protein monomers and domains, "with barely any space for further improvements at the backbone level except for very specific details, irregular secondary structures, and mutational effects that remain challenging to predict."[5] For prediction of protein oligomer assemblies, AlphaFold-based methods "show progress, though complex topologies and in particular antibody-antigen interactions are still difficult. Notably, a priori knowledge of stoichiometry significantly aids assembly prediction. Protein-ligand co-folding with AF3 demonstrated strong potential for pose prediction, outperforming many participants and some dedicated docking tools in baseline tests, but several caveats hold as discussed. Ligand affinity prediction is totally unreliable. Nucleic acid structure prediction lags considerably ...."[5]
2022: CASP 15
Overall, AlphaFold2 continued to "convincingly outperform all other methods" when various methods were compared using "fully automated mode with default parameter settings, without any manual interventions"[7]. AlphaFold2 predictions had a mean GDT-TS score of 73 (100 meaning perfect, and 0, meaningless). ESMFold, which is not based upon multiple sequence alignments, attained second best for backbone positioning (mean GDT-TS 61.6), outperforming RoseTTAFold (which is MSA based) for >80% of cases[7]. Individual domains were reliably predicted in the 19 multidomain targets, but predictions of domain orientations were less successful[7]. As an example, AlphaFold 2 achieved the best prediction for one large multi-domain target T1154, but the GDT-TS was only 24[7]. There is considerable room for improvement in prediction of side-chain positioning: while AlphaFold2 was most successful, its mean GDC-SC score fell short of 50[7]. Targets in CASP 15 (2022) included several new categories: 12 with RNA[8][9], some ligand protein complexes, and 41 quaternary assembly protein complexes[10]. "... for the vast majority of proteins and protein complexes, AlphaFold can produce a model close to experimental quality."[11]. The success rate for overall fold and interface prediction in complexes was 90%, vs. 31% in CASP 14[12]. This was "largely due to the incorporation of DeepMind's AF2-Multimer approach into custom-built prediction pipelines"[12].
2020: CASP 14
The best predictions at CASP 13 (2018) correctly predicted "folds" and the topology of secondary structure elements (helices and beta strands), but fell short of correctly predicting entire structures in detail.
In CASP 14 (2020), the AlphaFold2[13][14] system of DeepMind[15][16] demonstrated a major breakthrough[17][18][19][20]. AlphaFold2 was far better able, among over 100 competing groups, to predict structures, including sidechain positions, so close to the subsequently revealed X-ray crystallographic structures as to differ by little more than the differences between two independently-determined X-ray structures of the same molecule. It did this for about two-thirds of the targets in the competition. AlphaFold2 has been hailed as largely solving the protein structure prediction problem for single-chain proteins[17][18][19][20]. "Never in my life had I expected to see a scientific advance so rapid." said Mohammed AlQuraishi of Columbia University[17].
|
See AlphaFold2 examples from CASP 14 for some detailed comparisons. |
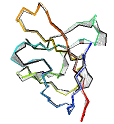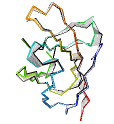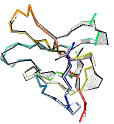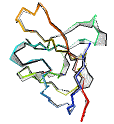  |
  |
Visit the DeepMind AlphaFold2 team and hear commentary by luminaries such as John Moult at YouTube. |
AlphaFold2 Methods
AlphaFold2 uses deep machine learning from the Protein Data Bank and sequence databases, and relies heavily on distances between beta-carbons[13], using co-evolution rates determined from multiple sequence alignments. AlphaFold2 is trained from data in the PDB to predict "the distances between pairs of residues, which convey more information about the structure than contact predictions."[13] By one estimate[21], "the DeepMind team had roughly two orders of magnitude more computational resources" than did academic groups competing in CASP 14. Further information about methods was provided by AlQuraishi[17].
CASP 14 Global Distance Test Results
Performance was judged overall, in large part, by the global distance test total score or GDT_TS[22][23]. GDT_TS values range from 0 (a meaningless prediction) to 100 (a perfect prediction; see Calculating GDT TS. "Random predictions give around 20; getting the gross topology right gets one to ~50; accurate topology is usually around 70; and when all the little bits and pieces, including side-chain conformations, are correct, GDT_TS begins to climb above 90."[17].
A GDT_TS value of ~90 means that the prediction is as close to an empirical model as would be an independently obtained second empirical model. GDT_TS gives an overall average measure of how close each amino acid in the predicted model is to those in the empirical model, taking into account many different superpositions of the two models. It is less sensitive to outlier regions than is the root mean square deviation (RMSD)[24]. "RMSD uses the actual distances between alpha carbons, where GDT works with the percentage of alpha carbons that are found within certain cutoff distances of each other."[25]
Based on GDT_TS, the most successful predictions were by AlphaFold2, which achieved a median GDT_TS of 92.4[17]. The second most successful predictions were by by BAKER (David Baker group). A group with median success was CAO-QA1 (Renzhi Cao, Kyle Hippe, & Mikhail Korovnik).
| Group Name | Rank | GDT_TS ≥ 90 | GDT_TS ≥ 87 | GDT_TS High | GDT_TS Median | GDT_TS Low |
|---|---|---|---|---|---|---|
| AlphaFold2 | 1 | 55% | 68% | 99 | 92 | 45 |
| BAKER | 2 | 5% | 8% | 96 | 70 | 25 |
| CAO-QA1 | 73 | 1% | 1% | 91 | 36 | 4 |
Each of the three groups in the above table submitted 92 predictions. Data are for FM (Free Modeling) and TBM (Template Based Modeling) targets[26].
CASP 14 Rankings
AlphaFold2 ranked first, by a wide margin, for all categories of targets. Groups making predictions were ranked by the sums of the Z-scores for their predictions[27]. A Z-score is the GDT_TS score for one prediction minus the mean of all GDT_TS scores for the target in question, divided by the standard deviation for all GDT_TS scores. For 92 single-domain targets, AlphaFold2's Z-score sum was 2.7 fold higher than the second best, which was the group of David Baker. It was 14-fold higher than the median. For 10 multi-domain targets, AlphaFold2's Z-score sum was 3.6-fold higher than the second best (again, the David Baker group), and 23-fold higher than the median.
AlphaFold2 Pros and Cons
In February, 2021, AlphaFold2 is not yet available to the general scientific community. When it, or another system based on the same principles, does become available, it is not clear how much computing power will be needed. By one estimate[21], "the DeepMind team had roughly two orders of magnitude more computational resources" than did academic groups competing in CASP 14.
AlphaFold2 was developed by DeepMind, a for-profit company whose parent company is Alphabet, Inc., the parent company of Google[16]. An ethical question arises, since AlphaFold2 was trained on public datasets, largely funded by public money[19]. "Big as DeepMind's war chest might be, the taxpayers' investment that has made their achievement possible is several orders of magnitude larger."[21].
When the new technology becomes widely available, X-ray crystallographers may often be able to skip solving the phase problem, since they can solve their diffraction data by molecular replacement, using predicted structures -- at least for single chain and single domain structures. This has already occurred: the group of Henning Tidow had toiled away for over a year on a structure which they were able to solve in less than a day using a prediction from DeepMind[21].
Neither empirical methods nor theoretical methods are obsolete.
- 1. AlphaFold2 does very well for about 2/3 of 92 single chain domains targeted in CASP 14, but less well for the remaining third. Its performance for sequence families not represented in CASP 14, and not well represented in the Protein Data Bank training set, remains to be seen.
- 2. AlphaFold2 does not predict interactions between chains[19] forming functional biological assemblies.
- 3. AlphaFold2 does not predict ligand binding, including the positions of metals in the one-third of proteins that are metalloproteins[19].
- 4. AlphaFold2 does not predict protein kinetics and allostery, often crucial for function[19].
- 5. AlphaFold2 does not predict the trajectory of how a protein folds, only the final structure[19].
AlphaFold Servers and Database
The methods and open-source code, as well as the advent of free servers offering to predict structures, and a huge database of predictions were published or became available in July, 2021. Please see AlphaFold.
2018: CASP 13
Excerpts from the conclusions: "... the ability of predicting hard protein folds at the tertiary level has increased enormously ..." "On the other hand, important global and local features of prediction models are still seldom as accurate as in the experimental structure. This is the case of enzyme active sites and ligand binding sites, where the predicted arrangement of the amino acids side chains involved in ligand binding and substrate specificity has not achieved the level of accuracy required to confidently infer their function .... Accurate prediction of loops is still a challenging task*. As they are often involved in protein interactions, their incorrect prediction can compromise the accuracy of the interacting surface and overall structure of the complex." "... the ability of current methods in modeling the correct quaternary structure of proteins remains rudimentary and shows little progress compared to what observed at the tertiary level."[28]
"The most recent experiment (CASP13 held in 2018) saw dramatic progress in structure modeling without use of structural templates (historically 'ab initio' modeling). Progress was driven by the successful application of deep learning techniques to predict inter-residue distances. In turn, these results drove dramatic improvements in three-dimensional structure accuracy: With the proviso that there are an adequate number of sequences known for the protein family, the new methods essentially solve the long-standing problem of predicting the fold topology of monomeric proteins."[29]
*Fig. 4 in Kryshtafovych et al.[29] illustrates how, in the case of 6cci (~350 residues), the core of the protein is well-predicted, while the surface loops are poorly predicted. Surfaces of folded proteins are generally critical in their functions.
2008: CASP 8
In CASP 8 (2008), there were 13 "template free" targets, that is, sequences for which no significant sequence identity occurred for any empirically solved entry in the PDB. These are the most difficult to predict, as they must be predicted by ab initio methods. 102 groups submitted predictions. Assessing the quality of a prediction is not simple, given that even "good" predictions can have high root mean square (RMS) deviations for alpha carbon alignment, e.g. due to a hinge[30]. Several assessment methods were used, each emphasizing different qualities. A number of groups submitted good predictions for six of the thirteen targets[30]. None of the submitted models was judged to be satisfactory for four of the thirteen targets[30].
2004: CASP 6
In 2004, for about one out of four cases of small domains of less than 85 amino acids, the best predictions were within about 1.5 Å (RMS for carbon alphas) of the true structure[31]. (Independent determinations of the same protein by empirical methods generally agree within <1.0 Å RMS for carbon alphas.)
See Also
- AlphaFold/Index, a list of pages in Proteopedia about Alphafold.
- Calculating GDT TS
- Theoretical models displayed in Proteopedia must be clearly identified: see Proteopedia:Policy#Theoretical Models using methods explained at Proteopedia:Cookbook#Theoretical Models.
- Category:Theoretical Model
- Homology modeling
- Practical Guide to Homology Modeling
- Homology modeling servers
- Structural genomics
- CASP
- CAMEO
- Folding@Home and Rosetta@home use distributed computing to fold proteins and address structural biology issues.
- Protein Folding Game called Foldit: Are humans good at folding? Can crowdsourcing help the folding problem? Game to assess how well you folded a solved protein. Structures given as folding puzzles aren't include solved structures and necessarily derived from theoretical; however, the goal of users is to produce a theoretical model that matches the known structure without referring to known structure. Check out an article on it in Nature[32] and this coverage of the project.
- Zhou and Robinson[33] discuss integrating mass spectrometry and structural models to understand location of post-translational modifications. They include examples of superimposing homologous proteins in a complex and superposing on that the location of modifications to better understand complexes and the interactions of modified portions of proteins in such complexes.
- RNaseA Nobel Prizes for discussion of protein folding.
- Biological Complexes
References & Links
- ↑ Raval A, Piana S, Eastwood MP, Dror RO, Shaw DE. Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins. 2012 Aug;80(8):2071-9. doi: 10.1002/prot.24098. Epub 2012 May 15. PMID:22513870 doi:10.1002/prot.24098
- ↑ Critical Assessment of techniques for protein Structure Prediction (CASP).
- ↑ Noivirt-Brik O, Prilusky J, Sussman JL. Assessment of disorder predictions in CASP8. Proteins. 2009 Aug 21. PMID:19774619 doi:10.1002/prot.22586
- ↑ CAPRI: Critical Assessment of PRediction of Interactions.
- ↑ 5.0 5.1 5.2 Abriata LA, Dal Peraro M. Practical Outcomes From CASP16 for Users in Need of Biomolecular Structure Prediction. Proteins. 2026 Jan;94(1):435-446. PMID:41088961 doi:10.1002/prot.70078
- ↑ Zhang J, Yuan R, Kryshtafovych A, Pei J, Kretsch RC, Schaeffer RD, Zhou J, Das R, Grishin NV, Cong Q. Assessment of Protein Complex Predictions in CASP16: Are We Making Progress? Proteins. 2026 Jan;94(1):106-130. PMID:41170922 doi:10.1002/prot.70068
- ↑ 7.0 7.1 7.2 7.3 7.4 Moussad B, Roche R, Bhattacharya D. The transformative power of transformers in protein structure prediction. Proc Natl Acad Sci U S A. 2023 Aug 8;120(32):e2303499120. PMID:37523536 doi:10.1073/pnas.2303499120
- ↑ Das R, Kretsch RC, Simpkin A, Mulvaney T, Pham P, Rangan R, Bu F, Keegan R, Topf M, Rigden D, Miao Z, Westhof E. Assessment of three-dimensional RNA structure prediction in CASP15. bioRxiv. 2023 Jul 17:2023.04.25.538330. PMID:37162955 doi:10.1101/2023.04.25.538330
- ↑ Kretsch RC, Andersen ES, Bujnicki JM, Chiu W, Das R, Luo B, Masquida B, McRae EKS, Schroeder GM, Su Z, Wedekind JE, Xu L, Zhang K, Zheludev IN, Moult J, Kryshtafovych A. RNA target highlights in CASP15: Evaluation of predicted models by structure providers. Proteins. 2023 Jul 19. PMID:37466021 doi:10.1002/prot.26550
- ↑ Kryshtafovych A, Antczak M, Szachniuk M, Zok T, Kretsch RC, Rangan R, Pham P, Das R, Robin X, Studer G, Durairaj J, Eberhardt J, Sweeney A, Topf M, Schwede T, Fidelis K, Moult J. New prediction categories in CASP15. Proteins. 2023 Jun 12. PMID:37306011 doi:10.1002/prot.26515
- ↑ Elofsson A. Progress at protein structure prediction, as seen in CASP15. Curr Opin Struct Biol. 2023 Jun;80:102594. PMID:37060758 doi:10.1016/j.sbi.2023.102594
- ↑ 12.0 12.1 Ozden B, Kryshtafovych A, Karaca E. The Impact of AI-Based Modeling on the Accuracy of Protein Assembly Prediction: Insights from CASP15. bioRxiv. 2023 Jul 11:2023.07.10.548341. PMID:37503072 doi:10.1101/2023.07.10.548341
- ↑ 13.0 13.1 13.2 Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Zidek A, Nelson AWR, Bridgland A, Penedones H, Petersen S, Simonyan K, Crossan S, Kohli P, Jones DT, Silver D, Kavukcuoglu K, Hassabis D. Improved protein structure prediction using potentials from deep learning. Nature. 2020 Jan;577(7792):706-710. doi: 10.1038/s41586-019-1923-7. Epub 2020 Jan, 15. PMID:31942072 doi:http://dx.doi.org/10.1038/s41586-019-1923-7
- ↑ AlphaFold at Wikipedia.
- ↑ AlphaFold: a solution to a 50-year-old grand challenge in biology, DeepMind Blog, November 30, 2020.
- ↑ 16.0 16.1 DeepMind at Wikipedia.
- ↑ 17.0 17.1 17.2 17.3 17.4 17.5 AlphaFold2 @ CASP14: “It feels like one’s child has left home.” by Mohammed AlQuraishi, December 8, 2020.
- ↑ 18.0 18.1 Artificial intelligence solution to a 50-year-old science challenge could ‘revolutionise’ medical research, CASP Press Release, November 30, 2020.
- ↑ 19.0 19.1 19.2 19.3 19.4 19.5 19.6 Callaway E. 'It will change everything': DeepMind's AI makes gigantic leap in solving protein structures. Nature. 2020 Dec;588(7837):203-204. doi: 10.1038/d41586-020-03348-4. PMID:33257889 doi:http://dx.doi.org/10.1038/d41586-020-03348-4
- ↑ 20.0 20.1 DeepMind and CASP14 by John R. Helliwell, International Union of Crystallography Newsletter, December 4, 2020.
- ↑ 21.0 21.1 21.2 21.3 CASP14: what Google DeepMind’s AlphaFold 2 really achieved, and what it means for protein folding, biology and bioinformatics, a blog post by Carlos Outeir al Rubiera, December 3, 2020.
- ↑ GDT description at the CASP website.
- ↑ Global distance test at Wikipedia.
- ↑ Root mean square deviation at Wikipedia.
- ↑ GDT in the Foldit Wiki.
- ↑ Data from CASP 14 "Table Browser". Caution: A maximum of 1,200 results are shown. To see all results for a given group, you must select that group alone. If you select all groups, only the subset of predictions with the highest GDT_TS scores is shown for the subset of groups listed.
- ↑ TS Analysis: Group performance based on combined z-scores for CASP 14 at PredictionCenter.Org.
- ↑ Lepore et al., in press in Proteins: Structure, Function, and Bioinformatics, 2019. DOI: 10.1002/prot.25805
- ↑ 29.0 29.1 Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical Assessment of Methods of Protein Structure Prediction (CASP) - Round XIII. Proteins. 2019 Oct 7. doi: 10.1002/prot.25823. PMID:31589781 doi:http://dx.doi.org/10.1002/prot.25823
- ↑ 30.0 30.1 30.2 Ben-David M, Noivirt-Brik O, Paz A, Prilusky J, Sussman JL, Levy Y. Assessment of CASP8 structure predictions for template free targets. Proteins. 2009 Aug 21. PMID:19774550 doi:10.1002/prot.22591
- ↑ Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005 Sep 16;309(5742):1868-71. PMID:16166519 doi:309/5742/1868
- ↑ Cooper S, Khatib F, Treuille A, Barbero J, Lee J, Beenen M, Leaver-Fay A, Baker D, Popovic Z, Players F. Predicting protein structures with a multiplayer online game. Nature. 2010 Aug 5;466(7307):756-60. PMID:20686574 doi:10.1038/nature09304
- ↑ Zhou M, Robinson CV. When proteomics meets structural biology. Trends Biochem Sci. 2010 Jun 3. PMID:20627589 doi:10.1016/j.tibs.2010.04.007
Acknowledgements
Eric Martz thanks Roman Sloutsky, Can Özden, Jeanne Hardy, Scott Garman, Thomas Sawyer, Katie Wahlbeck, Erik Nordquist, Nathaniel Kuzio (University of Massachusetts, Amherst) and Woody Sherman (Silicon Therapeutics) for introducing him to CASP 14 and AlphaFold2.
